

Profiling glycosphingolipid changes in mouse and human cellular models of lysosomal free sialic acid storage disorder
- PMID: 41280660
- PMCID: PMC12639585
- DOI: 10.1016/j.ymgmr.2025.101275
Profiling glycosphingolipid changes in mouse and human cellular models of lysosomal free sialic acid storage disorder
Abstract
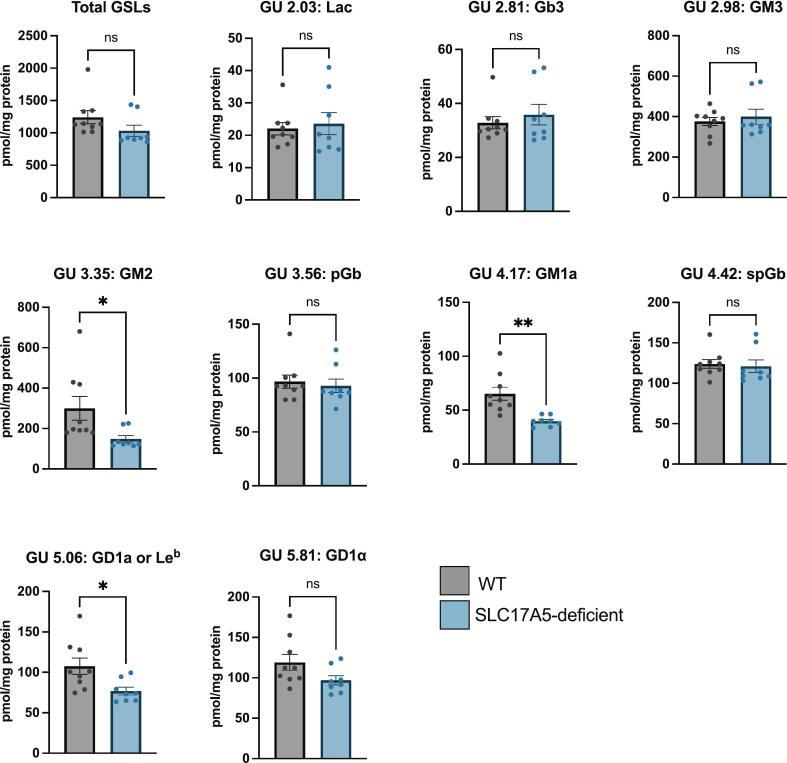
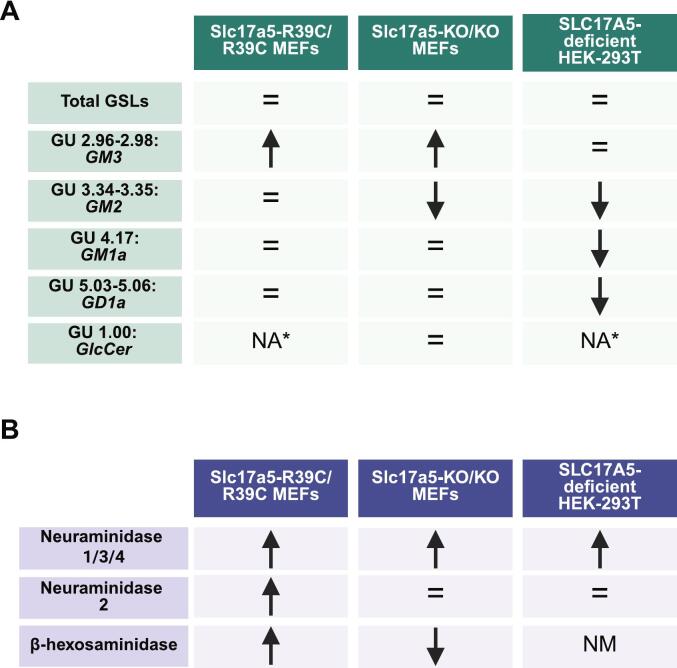
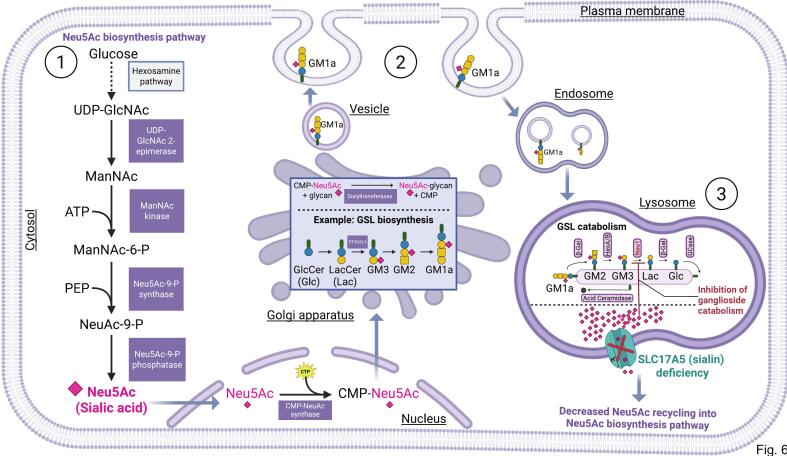
Free sialic acid storage disorder (FSASD) is an autosomal recessive lysosomal storage disease caused by biallelic pathogenic variants in SLC17A5, which encodes the lysosomal sialic acid transporter, sialin. FSASD is characterized by excessive lysosomal free sialic acid accumulation, leading to either a severe, early-onset lethal phenotype or a progressive neurodegenerative course. To characterize biochemical alterations in FSASD models, we performed comprehensive profiling of glycosphingolipids (GSLs), including sialylated species (i.e., gangliosides), in mouse embryonic fibroblasts (MEFs) derived from Slc17a5-R39C/R39C and Slc17a5-KO/KO mouse models, as well as in human SLC17A5-deficient HEK-293 T cells generated via CRISPR-Cas9-mediated non-homologous end joining. HPLC-based analyses demonstrated GM3 ganglioside accumulation in MEFs and significant reductions in a-series GSLs-including GM2, GM1a, and GD1a-in SLC17A5-deficient HEK-293 T cells. Analysis of neuraminidase 1/3/4 activities revealed consistently elevated activity across all cell models, while cytosolic neuraminidase 2 showed only a modest increase in Slc17a5-R39C/R39C MEFs. Preliminary quantification showed elevated free sialic acid across all models, consistent with the characteristic biochemical defect observed in FSASD and supporting their relevance for mechanistic studies. These findings highlight that free sialic acid storage leads to changes in GSL homeostasis in FSASD mouse (MEFs) and human (SLC17A5-deficient HEK-293T) cellular models, underscoring their utility as models for studying FSASD pathogenesis.
Keywords: Gangliosides; HEK-293 T; Mouse embryonic fibroblasts; Neuraminidase; Salla disease; Sialin.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures



References
-
- Verheijen F.W., et al. A new gene, encoding an anion transporter, is mutated in sialic acid storage diseases. Nat. Genet. 1999;23(4):462–465. - PubMed
-
- Adams D., Wasserstein M. In: GeneReviews((R)) Adam M.P., et al., editors. 2020. Free sialic acid storage disorders. Seattle (WA)
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
