

Proteome-wide model for human disease genetics
- PMID: 41286104
- PMCID: PMC12695638
- DOI: 10.1038/s41588-025-02400-1
Proteome-wide model for human disease genetics
Abstract
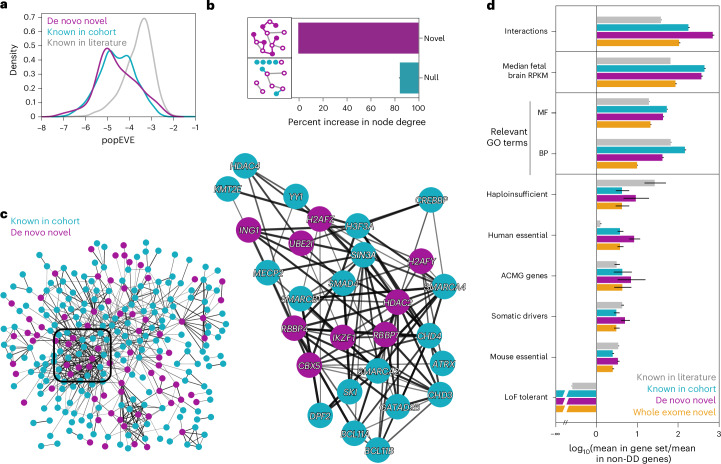
Missense variants remain a challenge in genetic interpretation owing to their subtle and context-dependent effects. Although current prediction models perform well in known disease genes, their scores are not calibrated across the proteome, limiting generalizability. To address this knowledge gap, we developed popEVE, a deep generative model combining evolutionary and human population data to estimate variant deleteriousness on a proteome-wide scale. popEVE achieves state-of-the-art performance without overestimating the burden of deleterious variants and identifies variants in 442 genes in a severe developmental disorder cohort, including 123 novel candidates. These genes are functionally similar to known disease genes, and their variants often localize to critical regions. Remarkably, popEVE can prioritize likely causal variants using only child exomes, enabling diagnosis even without parental sequencing. This work provides a generalizable framework for rare disease variant interpretation, especially in singleton cases, and demonstrates the utility of calibrated, evolution-informed scoring models for clinical genomics.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures















Update of
-
Proteome-wide model for human disease genetics.medRxiv [Preprint]. 2025 Mar 14:2023.11.27.23299062. doi: 10.1101/2023.11.27.23299062. medRxiv. 2025. Update in: Nat Genet. 2025 Dec;57(12):3165-3174. doi: 10.1038/s41588-025-02400-1. PMID: 38076790 Free PMC article. Updated. Preprint.
-
Deep generative modeling of the human proteome reveals over a hundred novel genes involved in rare genetic disorders.Res Sq [Preprint]. 2024 Jan 4:rs.3.rs-3740259. doi: 10.21203/rs.3.rs-3740259/v1. Res Sq. 2024. Update in: Nat Genet. 2025 Dec;57(12):3165-3174. doi: 10.1038/s41588-025-02400-1. PMID: 38260496 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
