

Spontaneous loss of T-cell tolerance to glutamic acid decarboxylase in murine insulin-dependent diabetes
- PMID: 7694152
- PMCID: PMC8216222
- DOI: 10.1038/366069a0
Spontaneous loss of T-cell tolerance to glutamic acid decarboxylase in murine insulin-dependent diabetes
Abstract
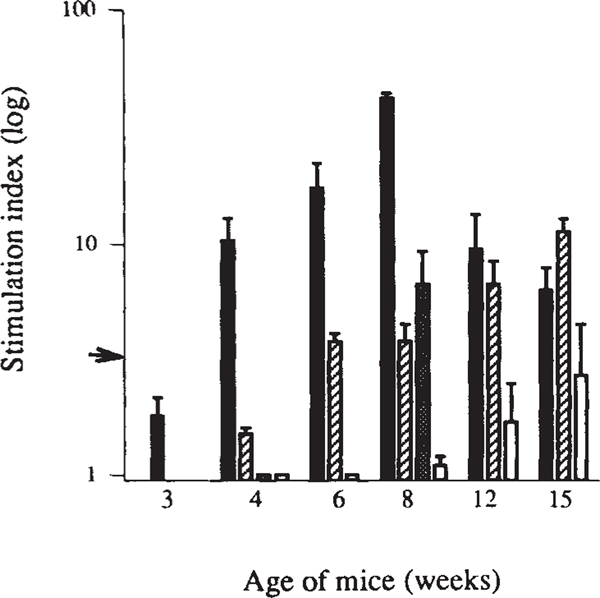
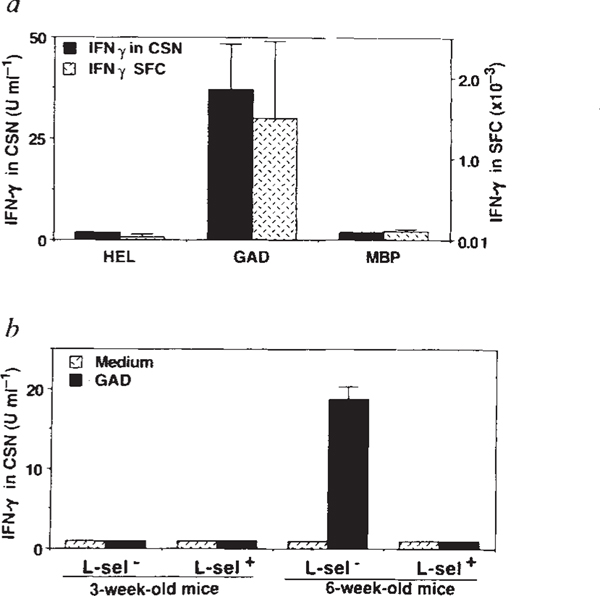
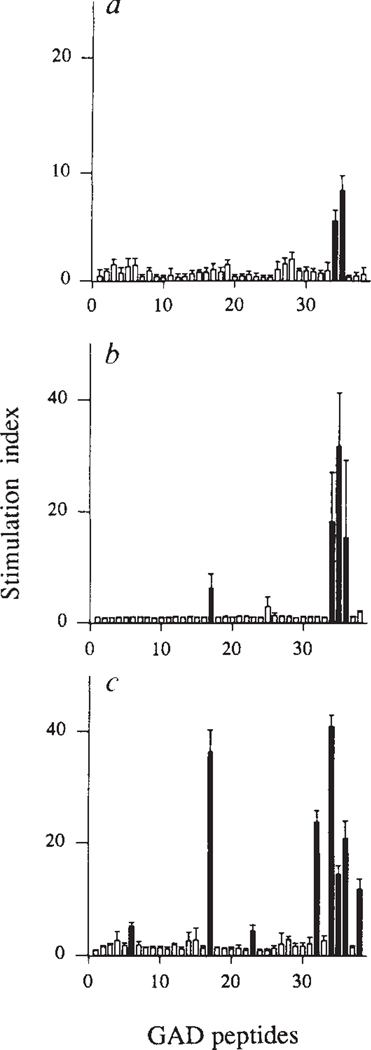
Insulin-dependent diabetes mellitus (IDDM) in non-obese diabetic (NOD) mice results from the T-lymphocyte-mediated destruction of the insulin-producing pancreatic beta-cells and serves as a model for human IDDM. Whereas a number of autoantibodies are associated with IDDM, it is unclear when and to what beta-cell antigens pathogenic T cells become activated during the disease process. We report here that a T-helper-1 (Th1) response to glutamate decarboxylase develops in NOD mice at the same time as the onset of insulitis. This response is initially limited to a confined region of glutamate decarboxylase, but later spreads intramolecularly to additional determinants. Subsequently, T-cell reactivity arises to other beta-cell antigens, consistent with intermolecular diversification of the response. Prevention of the spontaneous anti-glutamate decarboxylase response, by tolerization of glutamate decarboxylase-reactive T cells, blocks the development of T-cell autoimmunity to other beta-cell antigens, as well as insulitis and diabetes. Our data suggest that (1) glutamate decarboxylase is a key target antigen in the induction of murine IDDM; (2) autoimmunity to glutamate decarboxylase triggers T-cell responses to other beta-cell antigens, and (3) spontaneous autoimmune disease can be prevented by tolerization to the initiating target antigen.
Figures
Comment in
-
Diabetes. Spotlight on a neuronal enzyme.Nature. 1993 Nov 4;366(6450):15-7. doi: 10.1038/366015a0. Nature. 1993. PMID: 8232529 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
