

Voltage-independent calcium channels mediate slow oscillations of cytosolic calcium that are glucose dependent in pancreatic beta-cells
- PMID: 8013370
- PMCID: PMC2922863
- DOI: 10.1210/endo.135.1.8013370
Voltage-independent calcium channels mediate slow oscillations of cytosolic calcium that are glucose dependent in pancreatic beta-cells
Abstract
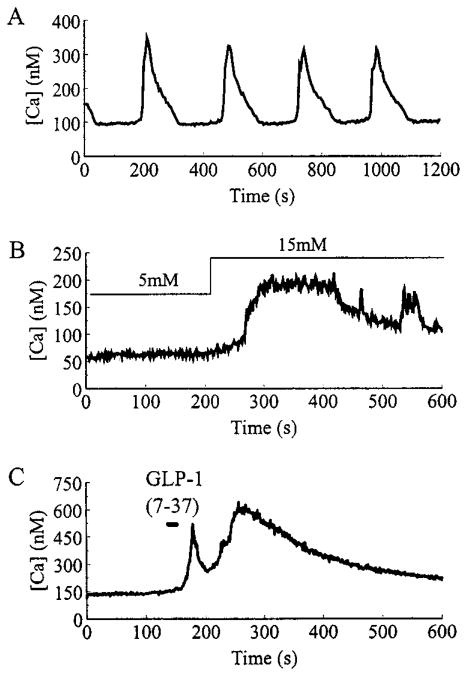
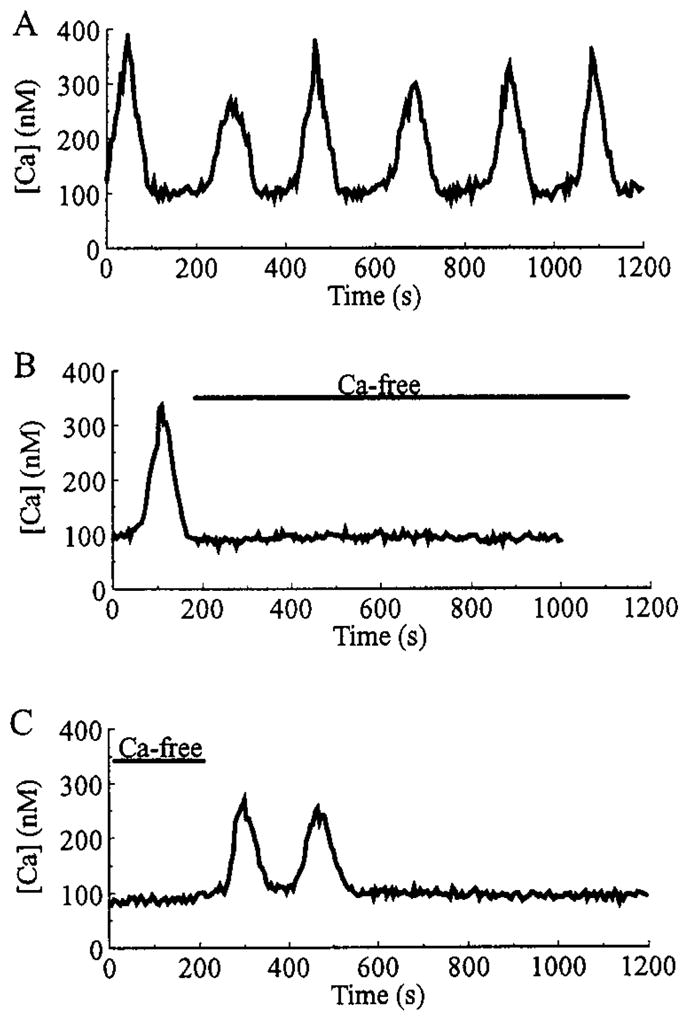
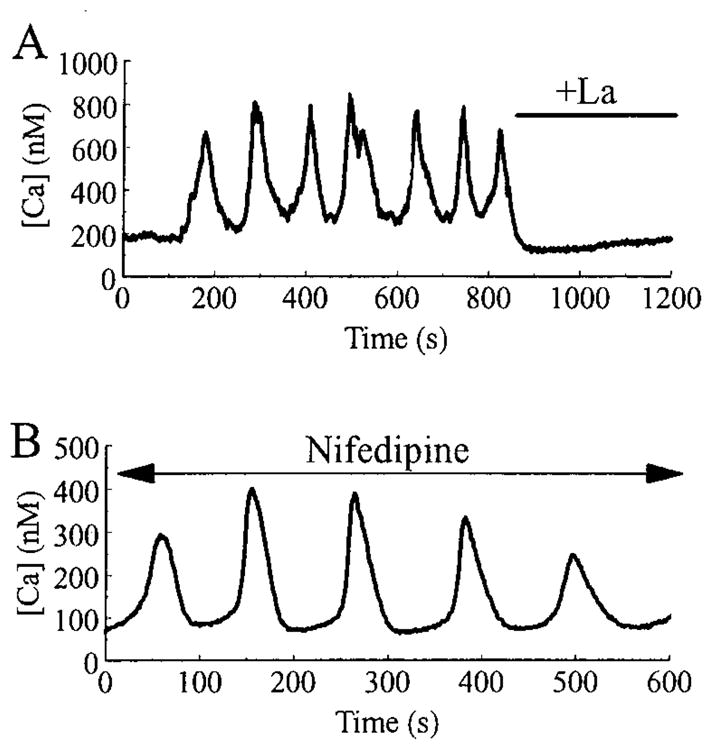
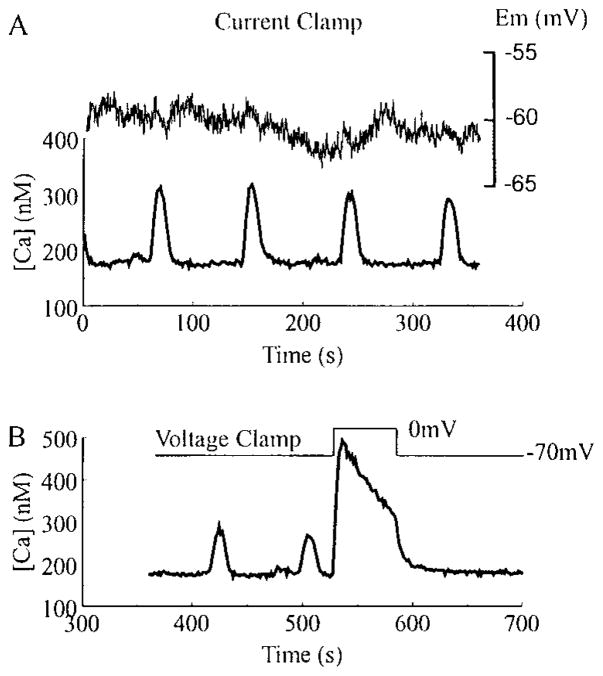
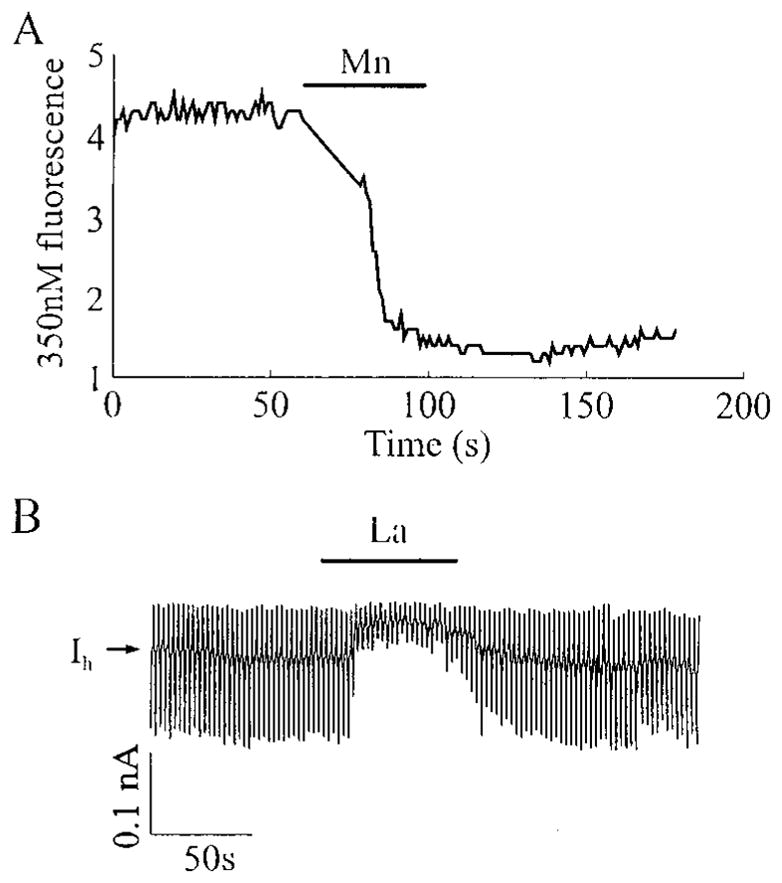
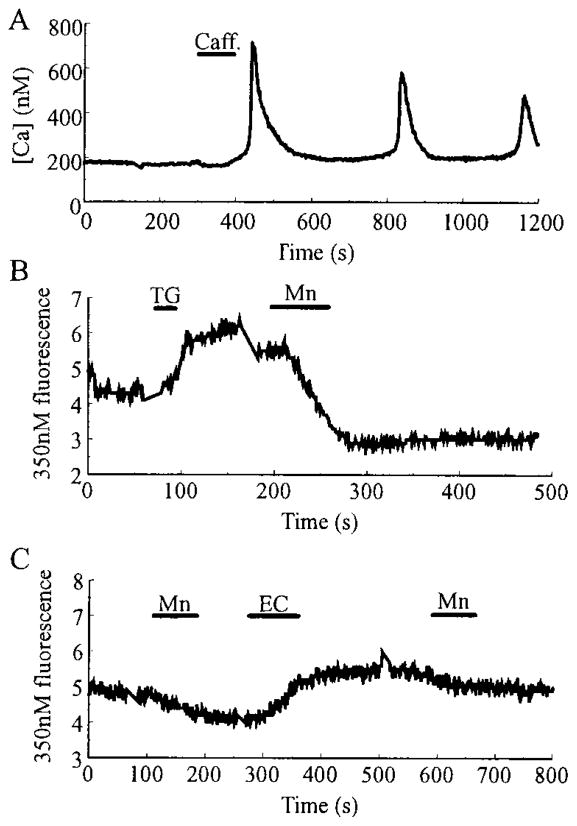
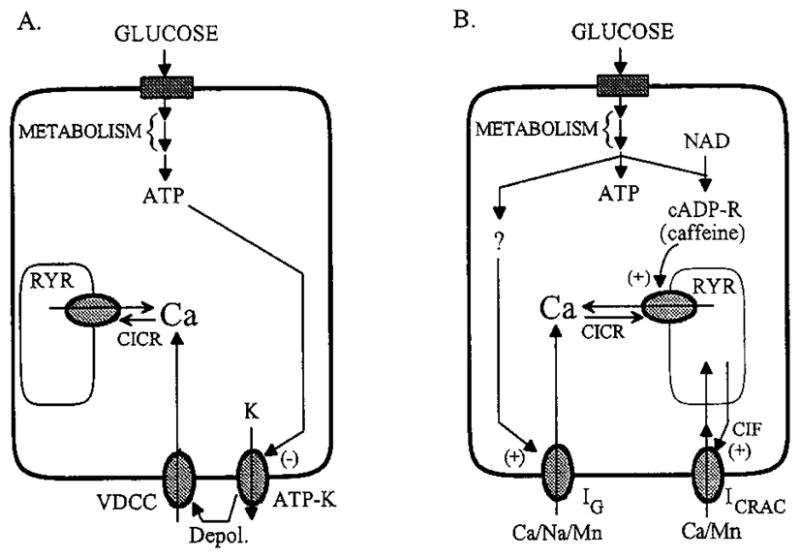
Pancreatic beta-cells and HIT-T15 cells exhibit oscillations of cytosolic calcium ([Ca2+]i) that are dependent on glucose metabolism and appear to trigger pulsatile insulin secretion. Significantly, differences in the pattern of this [Ca2+]i oscillatory activity may have important implications for our understanding of how glucose homeostasis is achieved during the feeding and fasting states. When single beta-cells are exposed to a stepwise increase in glucose concentration that mimics the transition from fasting to feeding states, fast irregular oscillations of [Ca2+]i are observed. Alternatively, when single beta-cells are equilibrated in a steady state concentration of glucose that mimics the fasting state, slow periodic oscillations of [Ca2+]i are noted. Here we report a fundamental difference in the mechanism by which glucose induces these two types of [Ca2+]i oscillatory activity. In agreement with previous studies, we substantiate a role for L-type voltage-dependent Ca2+ channels as mediators of the fast oscillations of [Ca2+]i observed after a stepwise increase in glucose concentration. In marked contrast, we report that voltage-independent calcium channels (VICCs) mediate slow oscillations of [Ca2+]i that occur when beta-cells are equilibrated in steady state concentrations of glucose. Slow [Ca2+]i oscillations are mediated by VICCs which are pharmacologically and biophysically distinguishable from voltage-dependent Ca2+ channels that mediate fast oscillations. Specifically, slow [Ca2+]i oscillations are blocked by extracellular La3+, but not by nifedipine, and are independent of changes in membrane potential. Measurement of membrane conductance also indicate an important role for VICCs, as demonstrated by a steady state inward Ca2+ current that is blocked by La3+. The steady state Ca2+ current appears to generate slow [Ca2+]i oscillations by triggering Ca(2+)-induced Ca2+ release from intracellular Ca2+ stores, a process that is mimicked by extracellular application of caffeine, a sensitizer of the ryanodine receptor/Ca2+ release channel. Depletion of intracellular Ca2+ stores with thapsigargin stimulated Mn2+ influx, suggesting the presence of Ca(2+)-release-activated Ca2+ channels. Taken together, these observations are consistent with a role for VICCs (possibly G-type channels) and/or Ca(2+)-release-activated Ca2+ channels as mediators of slow [Ca2+]i oscillations in beta-cells. We propose that slow oscillations of [Ca2+]i probably serve as important initiators of insulin secretion under conditions in which tight control of glucose homeostasis is necessary, as is the case during the fasting normoglycemic state.
Figures
References
-
- Corkey BE, Deeney JT, Glennon MC, Matschinsky FM, Prentki M. Regulation of steady-state free Ca2+ levels by the ATP/ADP ratio and orthophosphate in permeabilized RINm5F cells. J Biol Chem. 1988;263:4247–4253. - PubMed
-
- Woods NM, Cuthbertson KSR, Cobbold PH. Repetitive transient rises in cytoplasmic free calcium in hormone-stimulated hepatocytes. Nature. 1986;319:600–602. - PubMed
-
- Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. - PubMed
-
- Schlegal W, Winiger BP, Mollard P, Vacher P, Wuarin F, Zahnd GR, Wollheim CB, Dufy B. Oscillations of cytosolic Ca2+ in pituitary cells due to action potentials. Nature. 1987;329:719–721. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
