

Mitochondrial dysfunction is a primary event in glutamate neurotoxicity
- PMID: 8815895
- PMCID: PMC6579180
- DOI: 10.1523/JNEUROSCI.16-19-06125.1996
Mitochondrial dysfunction is a primary event in glutamate neurotoxicity
Abstract
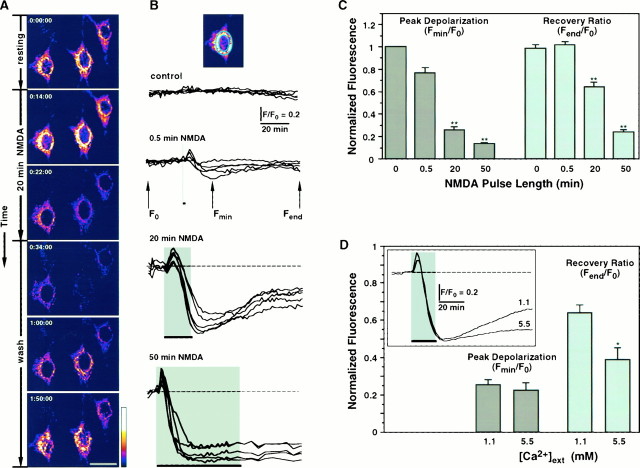
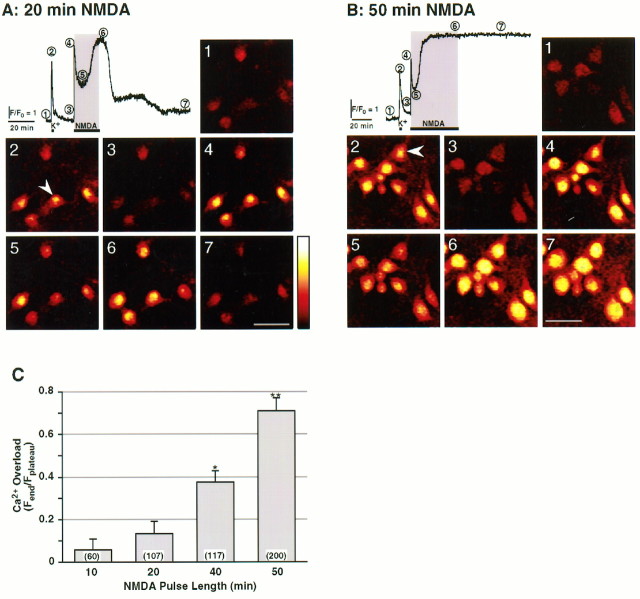
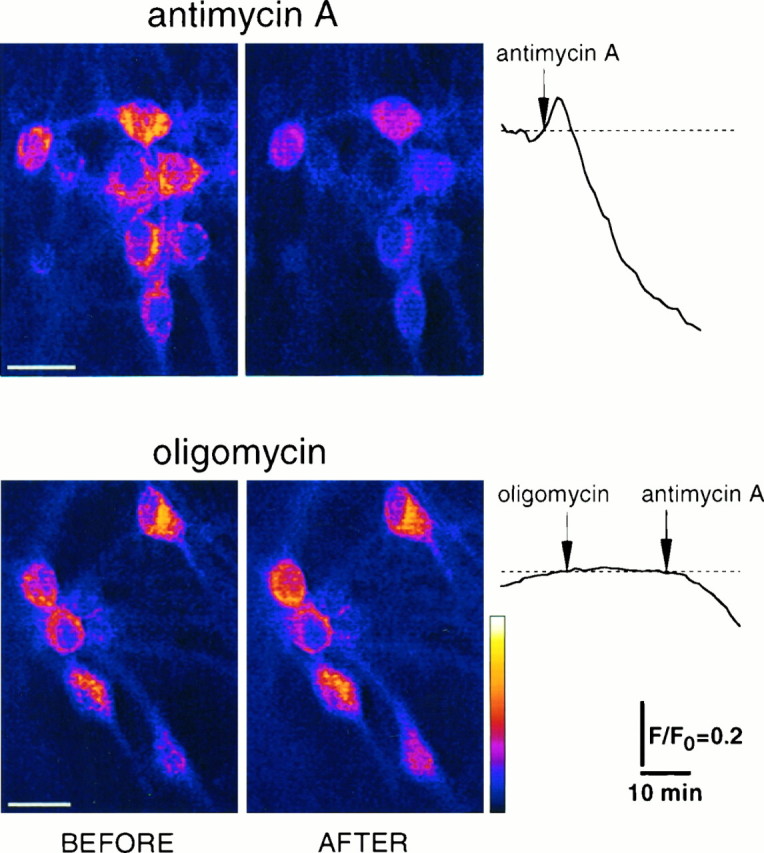
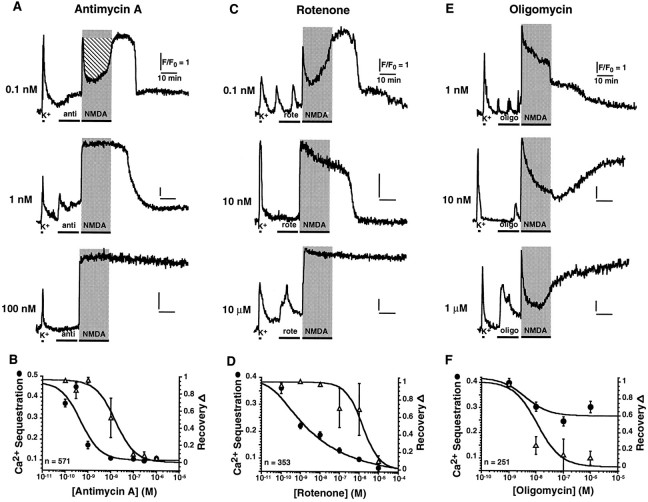
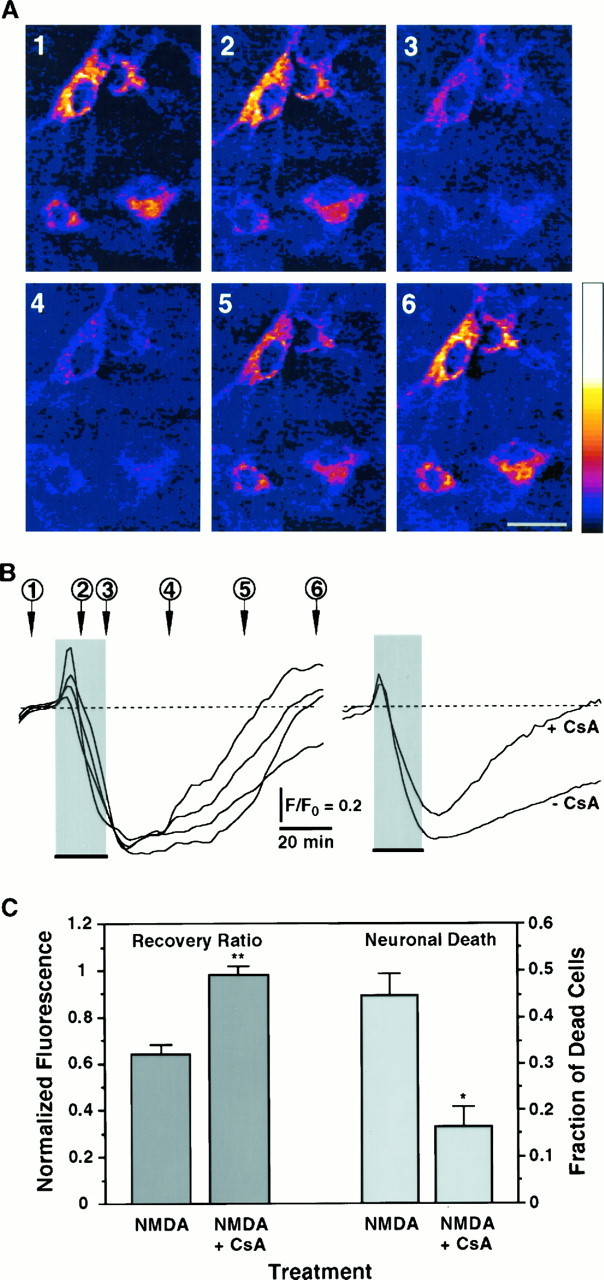
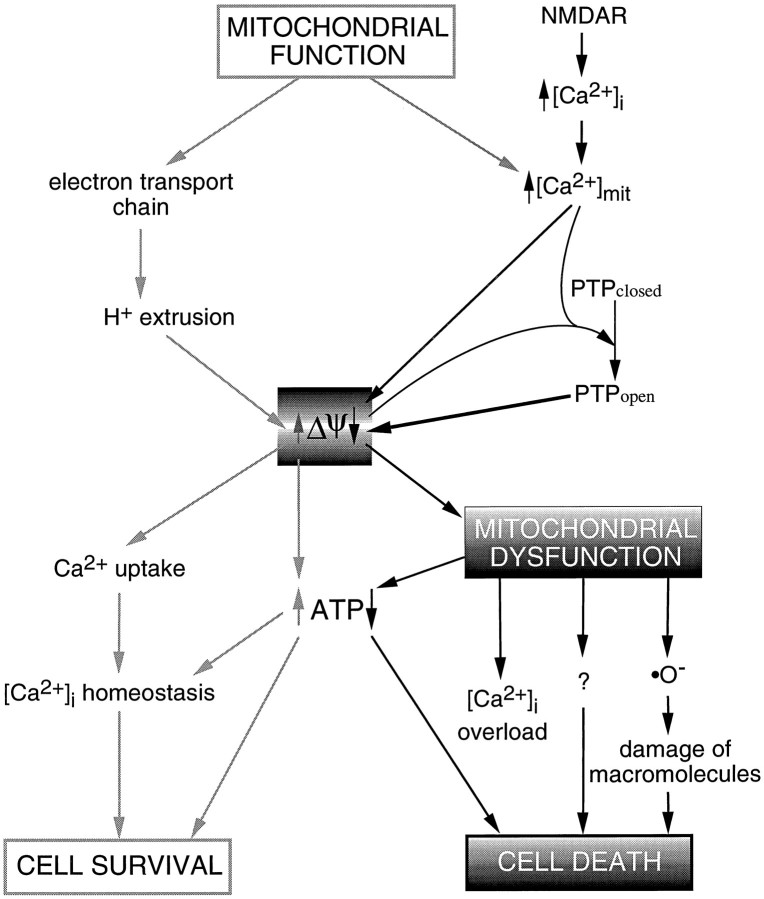
Excitotoxic neuronal death, associated with neurodegenerative disorders and hypoxic insults, results from excessive exposure to excitatory neurotransmitters. Glutamate neurotoxicity is triggered primarily by massive Ca2+ influx arising from overstimulation of the NMDA subtype of glutamate receptors. The underlying mechanisms, however, remain elusive. We have tested the hypothesis that mitochondria are primary targets in excitotoxicity by confocal imaging of intracellular Ca2+ ([Ca2+]i) and mitochondrial membrane potential (delta psi) on cultured rat hippocampal neurons. Sustained activation of NMDA receptors (20 min) elicits reversible elevation of [Ca2+]i. Longer activation (50 min) renders elevation of [Ca2+]i irreversible (Ca2+ overload). Susceptibility to NMDA-induced Ca2+ overload is increased when the 20 min stimuli are applied to neurons pretreated with electron transport chain inhibitors, thereby implicating mitochondria in [Ca2+]i homeostasis during excitotoxic challenges. Remarkably, delta psi exhibits prominent and persistent depolarization in response to NMDA, which closely parallels the incidence of neuronal death. Blockade of the mitochondrial permeability transition pore by cyclosporin A allows complete recovery of delta psi and prevents cell death. These results suggest that early mitochondrial damage plays a key role in induction of glutamate neurotoxicity.
Figures
References
-
- Akerman KE. Changes in membrane potential during calcium ion influx and efflux across the mitochondrial membrane. Biochim Biophys Acta. 1978;502:359–366. - PubMed
-
- Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. - PubMed
-
- Beal MF. Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative illnesses? Ann Neurol. 1992;31:119–130. - PubMed
-
- Beatrice MC, Palmer JW, Pfeiffer DR. The relationship between mitochondrial membrane permeability, membrane potential, and the retention of Ca2+ by mitochondria. J Biol Chem. 1980;255:8663–8671. - PubMed
-
- Bernardi P, Broekemeier KM, Pfeiffer DR. Recent progress on regulation of the mitochondrial permeability transition pore, a cyclosporin-sensitive pore in the inner mitochondrial membrane. J Bioenerg Biomembr. 1994;26:509–517. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous