

Amyloid beta peptide of Alzheimer's disease downregulates Bcl-2 and upregulates bax expression in human neurons
- PMID: 8922409
- PMCID: PMC6579094
- DOI: 10.1523/JNEUROSCI.16-23-07533.1996
Amyloid beta peptide of Alzheimer's disease downregulates Bcl-2 and upregulates bax expression in human neurons
Abstract
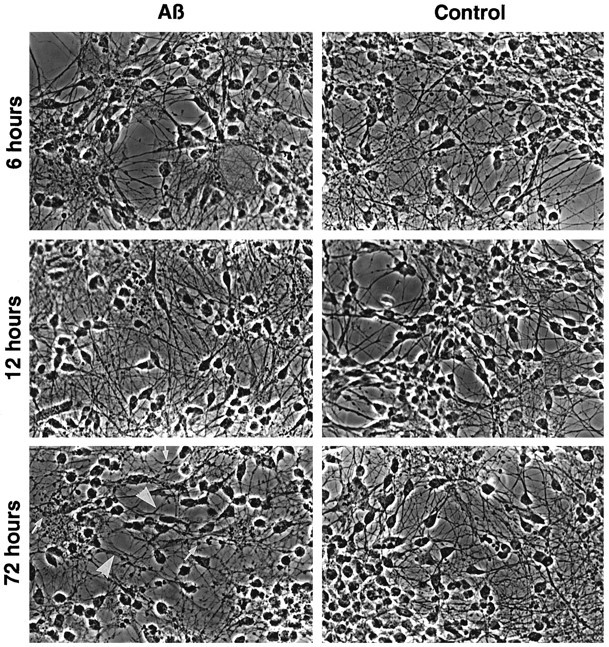
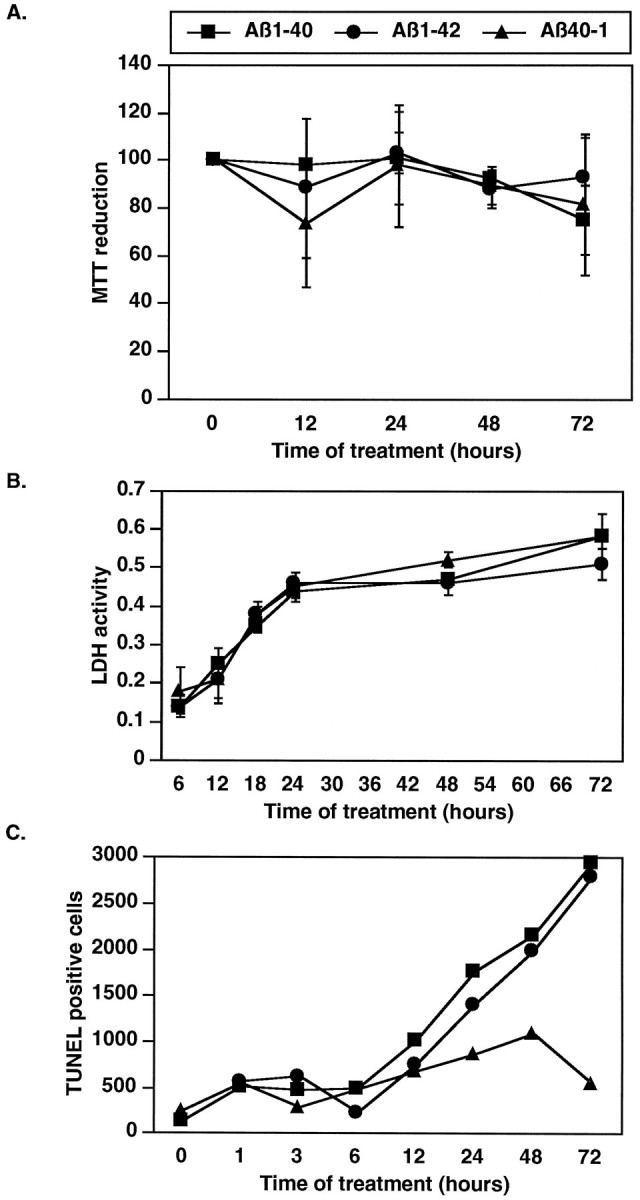
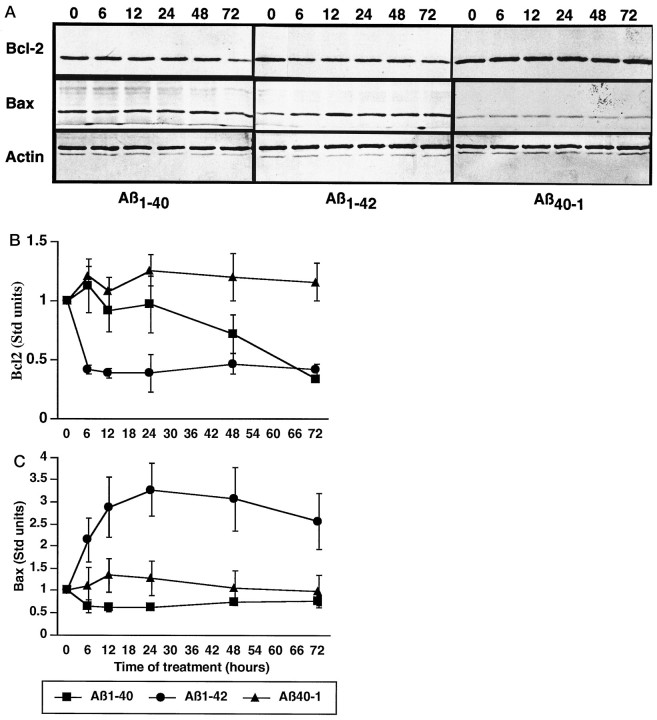
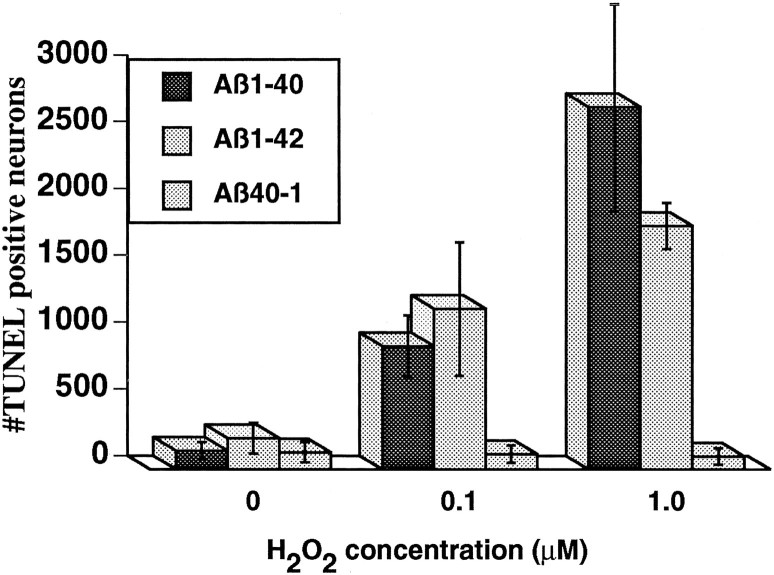
Neuronal apoptosis is a suspected cause of neurodegeneration in Alzheimer's disease (AD). Increased levels of amyloid beta peptide (Abeta) induce neuronal apoptosis in vitro and in vivo. The underlying molecular mechanism of Abeta neurotoxicity is not clear. The normal concentration of Abeta in cerebrospinal fluid is 4 nM. We treated human neuron primary cultures with 100 nM amyloid beta peptides Abeta(1-40) and Abeta(1-42) and the control reverse peptide Abeta(40-1). We find that although little neuronal apoptosis is induced by either peptide after 3 d of treatment, Abeta(1-42) provokes a rapid and sustained downregulation of a key anti-apoptotic protein, bcl-2, whereas it increases levels of bax, a protein known to promote cell death. In contrast, the Abeta(1-40) downregulation of bcl-2 is gradual, although the levels are equivalent to those of Abeta(1-42)-treated neurons by 72 hr of treatment. Abeta(1-40) does not upregulate bax levels. The control, reverse peptide Abeta(40-1), does not affect either bcl-2 or bax protein levels. In addition, we found that the Abeta(1-40)- and Abeta(1-42)- but not Abeta(40-1)-treated neurons had increased vulnerability to low levels of oxidative stress. Therefore, we propose that although high physiological amounts of Abeta are not sufficient to induce apoptosis, Abeta depletes the neurons of one of its anti-apoptotic mechanisms. We hypothesize that increased Abeta in individuals renders the neurons vulnerable to age-dependent stress and neurodegeneration.
Figures
References
-
- Anderson AJ, Cummings BJ, Cotman CW. Increased immunoreactivity of jun- and fos-related proteins in Alzheimer’s disease: association with pathology. Exp Neurol. 1994;125:286–295. - PubMed
-
- Bafy G, Miyashita J, Williamson J, Reed J. Apoptosis induced by withdrawal of interleukin-3 (IL-3)-dependent hemapoeitic cell line is associated with repartitioning of intracellular calcium and is blocked by enforced Bcl-2 oncoprotein production. J Biol Chem. 1993;268:6511–6519. - PubMed
-
- Behl C, Davis J, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid β protein toxicity. Cell. 1994;77:817–827. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials