

Defective subunit assembly underlies a digenic form of retinitis pigmentosa linked to mutations in peripherin/rds and rom-1
- PMID: 8943002
- PMCID: PMC19405
- DOI: 10.1073/pnas.93.24.13726
Defective subunit assembly underlies a digenic form of retinitis pigmentosa linked to mutations in peripherin/rds and rom-1
Abstract
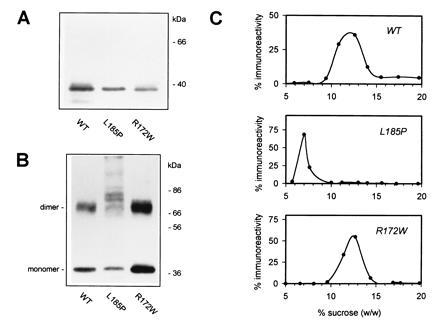
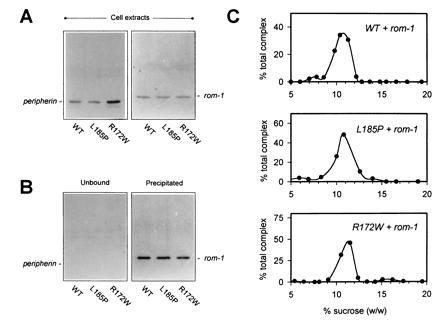
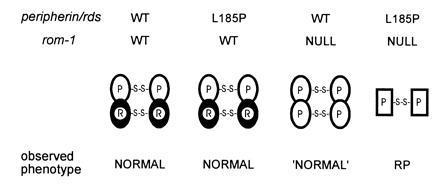
Retinitis pigmentosa (RP) is a group of progressive retinal dystrophies that include the most common hereditary degenerative disease affecting the retina. Although most disease phenotypes appear to result from defects at single genetic loci (monogenic), at least one instance of RP appears to require a coinheritance of defects in the unlinked peripherin/rds and rom-1 alleles (digenic), which encode the polypeptide subunits of an oligomeric transmembrane protein complex present at photoreceptor outer segment disc rims. Sedimentation velocity analysis was performed upon the affected gene products expressed heterologously in COS-1 cells to examine the assembly of the subunit polypeptides. The results indicate that the missense peripherin/rds mutant, L185P, which segregates with instance of digenically inherited RP, is conditionally defective with respect to its subunit assembly. Unlike wild-type peripherin/rds, the L185P mutant does not form native-like homotetramers on its own; however, the L185P mutant can assemble with wild-type rom-1 to form a structurally normal heterotetrameric complex. These findings provide a novel molecular-based rationale for the unusual digenic disease inheritance pattern and offer insight into regions of peripherin/rds and rom-1, which contribute to subunit-subunit interactions.
Figures
References
-
- Heckenlively J R. Retinitis Pigmentosa. Philadelphia: Lippincott; 1988.
-
- Berson E L. Invest Ophthalmol Vis Sci. 1993;34:1659–1676. - PubMed
-
- Shastry B S. Am J Med Genet. 1994;52:467–474. - PubMed
-
- Daiger S P, Sullivan L S, Rodriguez J A. Behav Br Sci. 1995;18:452–467.
-
- Dryja T P, Li T. Hum Mol Genet. 1995;4:1739–1743. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
