

Three-dimensional structure of human electron transfer flavoprotein to 2.1-A resolution
- PMID: 8962055
- PMCID: PMC26136
- DOI: 10.1073/pnas.93.25.14355
Three-dimensional structure of human electron transfer flavoprotein to 2.1-A resolution
Abstract
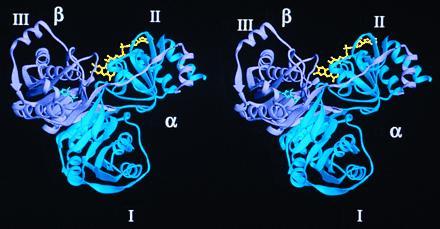
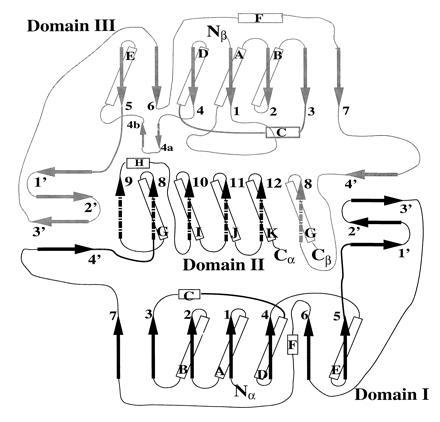
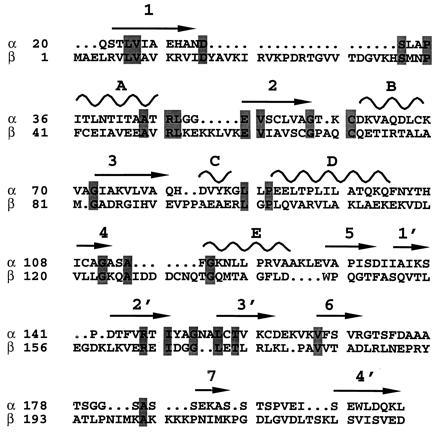
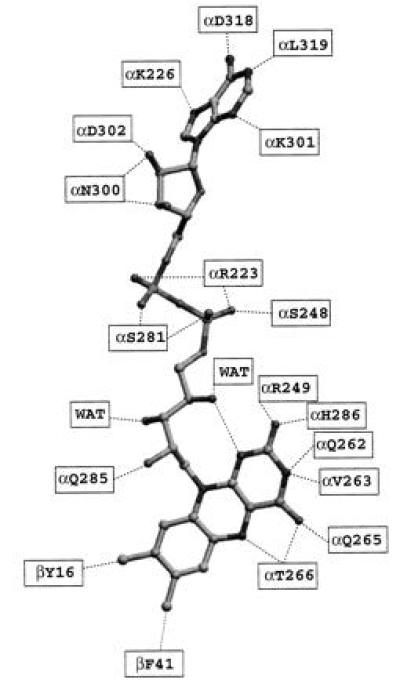
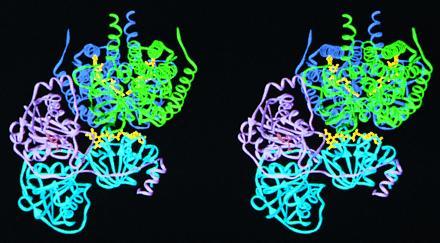
Mammalian electron transfer flavoproteins (ETF) are heterodimers containing a single equivalent of flavin adenine dinucleotide (FAD). They function as electron shuttles between primary flavoprotein dehydrogenases involved in mitochondrial fatty acid and amino acid catabolism and the membrane-bound electron transfer flavoprotein ubiquinone oxidoreductase. The structure of human ETF solved to 2.1-A resolution reveals that the ETF molecule is comprised of three distinct domains: two domains are contributed by the alpha subunit and the third domain is made up entirely by the beta subunit. The N-terminal portion of the alpha subunit and the majority of the beta subunit have identical polypeptide folds, in the absence of any sequence homology. FAD lies in a cleft between the two subunits, with most of the FAD molecule residing in the C-terminal portion of the alpha subunit. Alignment of all the known sequences for the ETF alpha subunits together with the putative FixB gene product shows that the residues directly involved in FAD binding are conserved. A hydrogen bond is formed between the N5 of the FAD isoalloxazine ring and the hydroxyl side chain of alpha T266, suggesting why the pathogenic mutation, alpha T266M, affects ETF activity in patients with glutaric acidemia type II. Hydrogen bonds between the 4'-hydroxyl of the ribityl chain of FAD and N1 of the isoalloxazine ring, and between alpha H286 and the C2-carbonyl oxygen of the isoalloxazine ring, may play a role in the stabilization of the anionic semiquinone. With the known structure of medium chain acyl-CoA dehydrogenase, we hypothesize a possible structure for docking the two proteins.
Figures

References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
