

High-resolution functional mapping of a cloned gene by genetic footprinting
- PMID: 9037048
- PMCID: PMC19786
- DOI: 10.1073/pnas.94.4.1304
High-resolution functional mapping of a cloned gene by genetic footprinting
Abstract
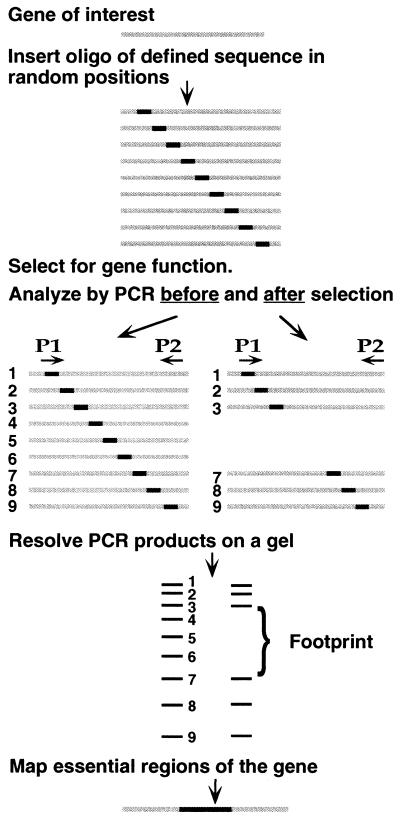
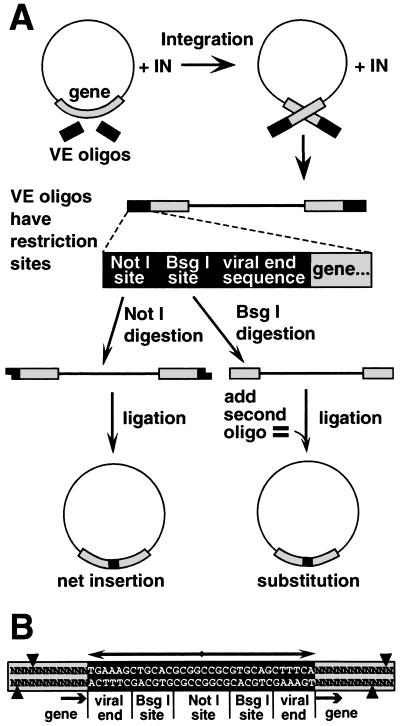
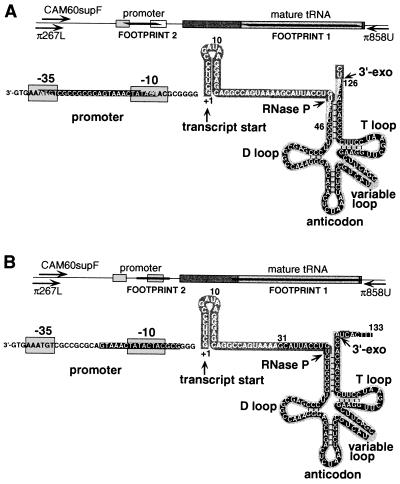
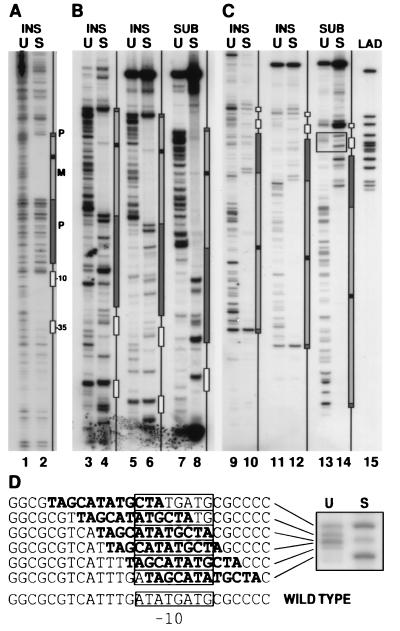
We describe an efficient method for introducing and analyzing a comprehensive set of mutations in a cloned gene to map its functional organization. The technique, genetic footprinting, uses a retroviral integrase to generate a comprehensive library of mutants, each of which bears a single insertion of a defined oligonucleotide at a random position in the gene of interest. This mutant library is selected for gene function en masse. DNA samples are isolated from the library both before and after selection, and the mutations represented in each sample are then analyzed. The analysis is designed so that a mutation at a particular location gives rise to an electrophoretic band of discrete mobility. For the whole library, this results in a ladder of bands, each band representing a specific mutation. Mutants in which the inserted sequence disrupts a feature that is required for the selected function, ipso facto, fail the selection. The corresponding bands are therefore absent from the ladder of bands obtained from the library after selection, giving rise to a footprint representing features of the gene that are essential for the selected function. Because the sequence of the inserted oligonucleotide is known, and its position can be inferred precisely from the electrophoretic mobility of the corresponding band, the precise location and sequence of mutations that disrupt gene function can be determined without isolating or sequencing individual mutants. This method should be generally applicable for saturation mutagenesis and high-resolution functional mapping of cloned DNA sequences.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
