

Contribution of sialic acid to the voltage dependence of sodium channel gating. A possible electrostatic mechanism
- PMID: 9089440
- PMCID: PMC2217074
- DOI: 10.1085/jgp.109.3.327
Contribution of sialic acid to the voltage dependence of sodium channel gating. A possible electrostatic mechanism
Abstract
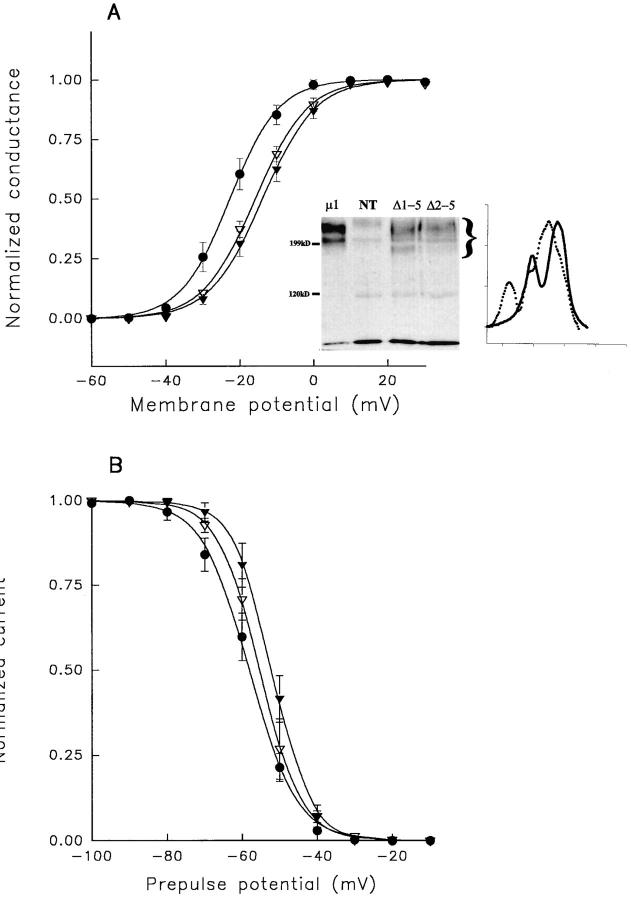
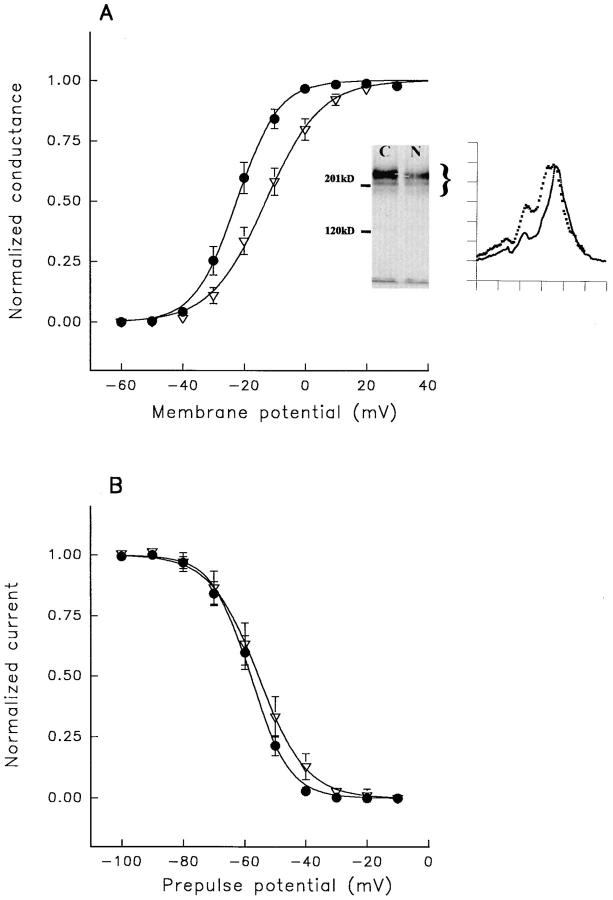
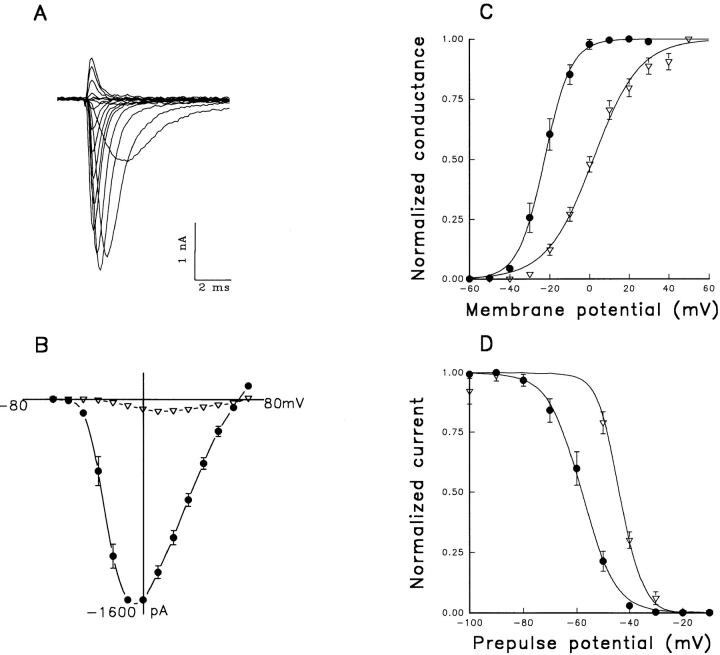
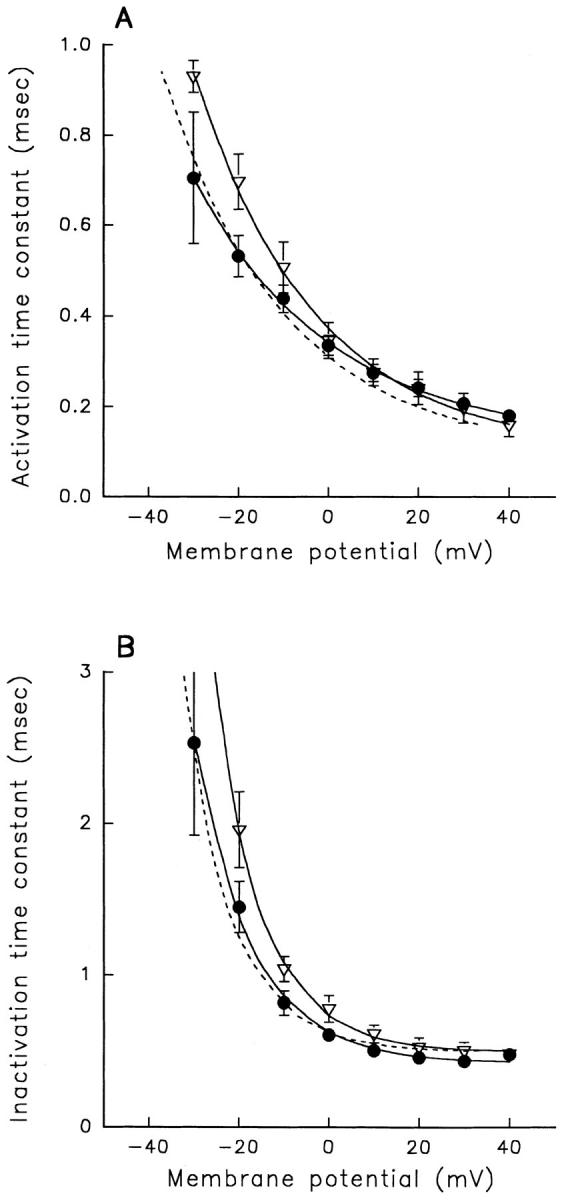
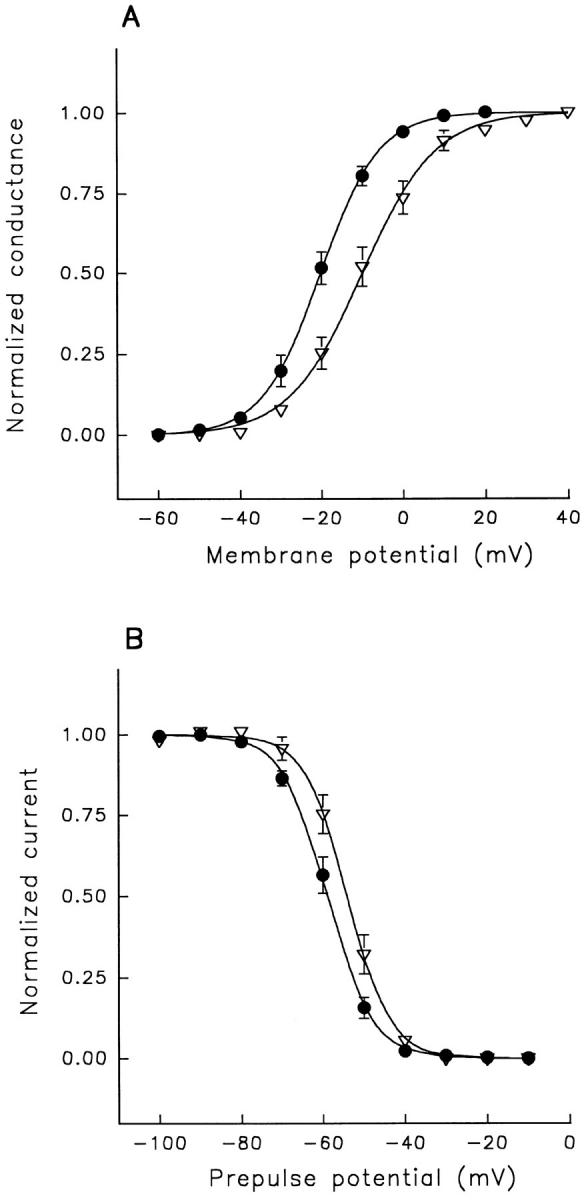
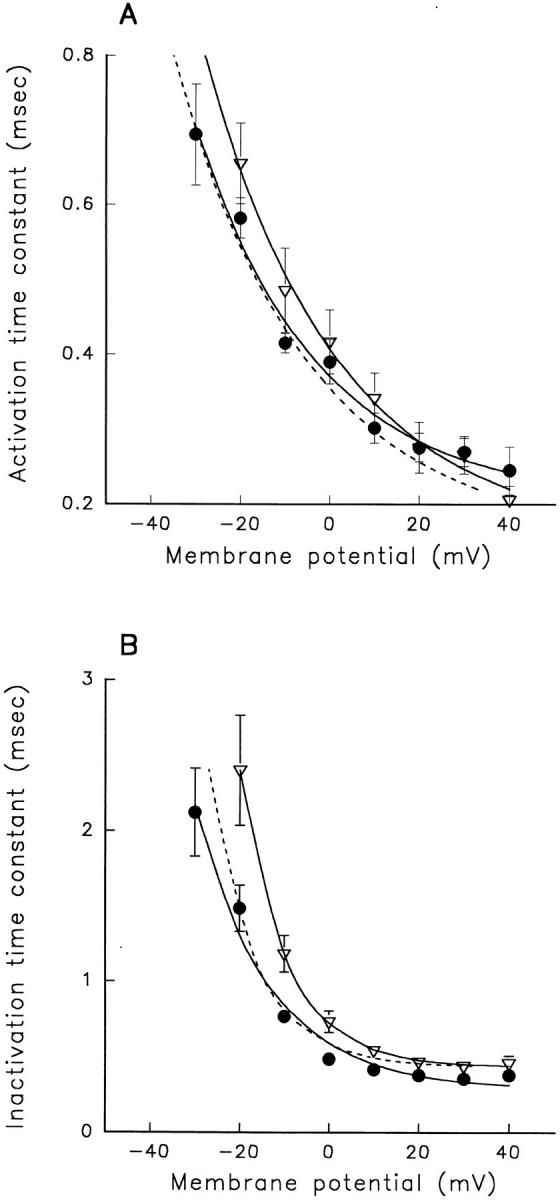
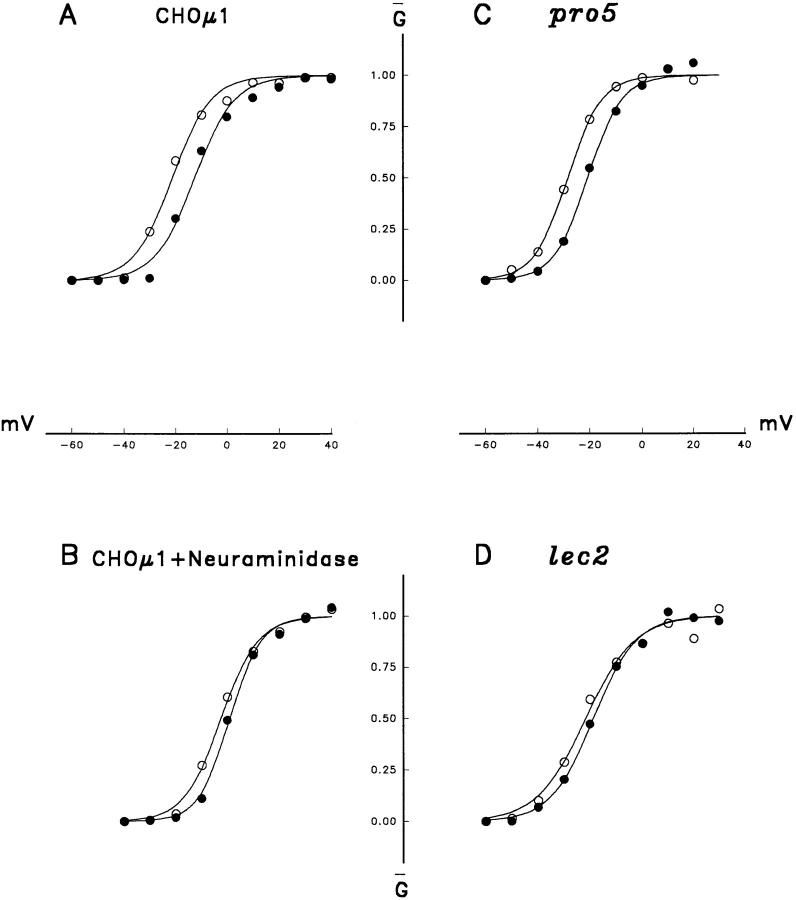
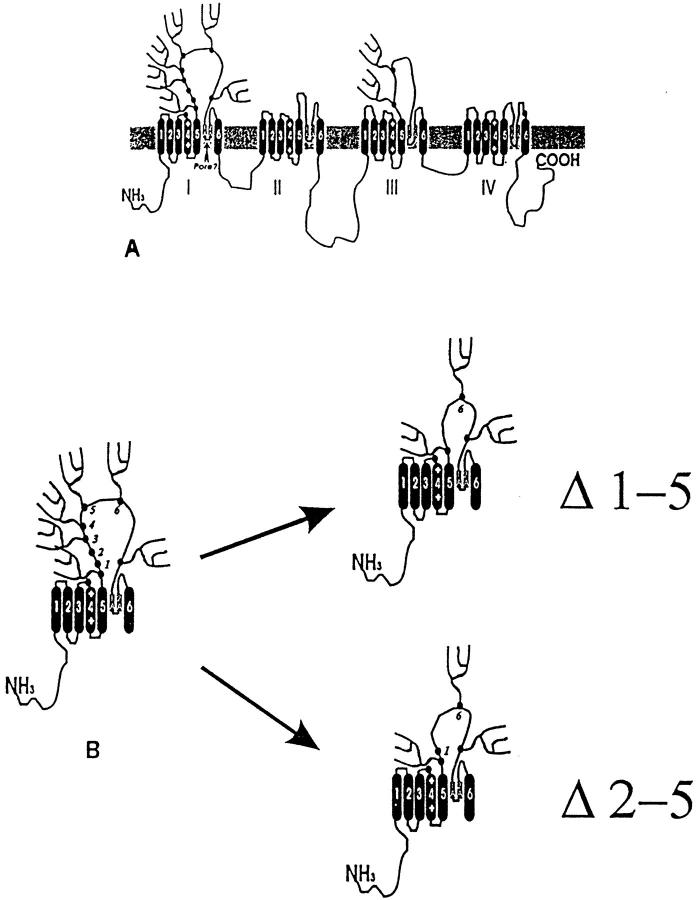
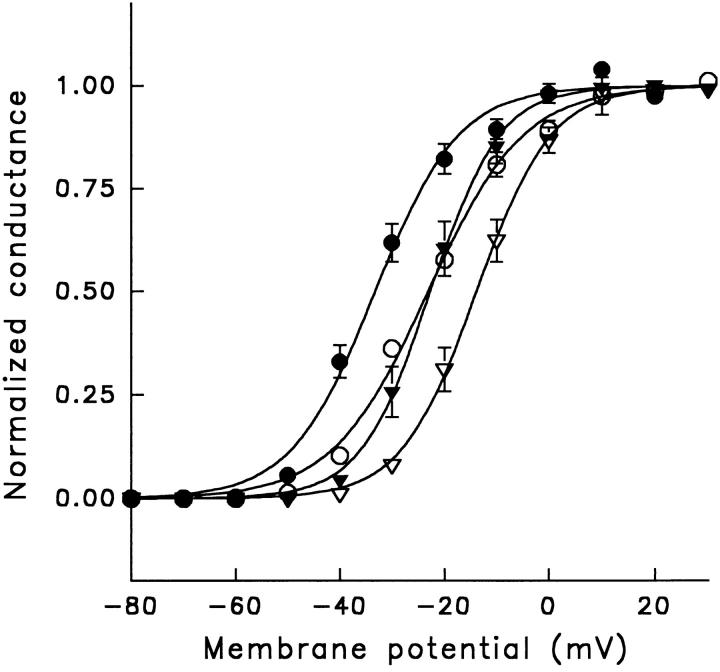
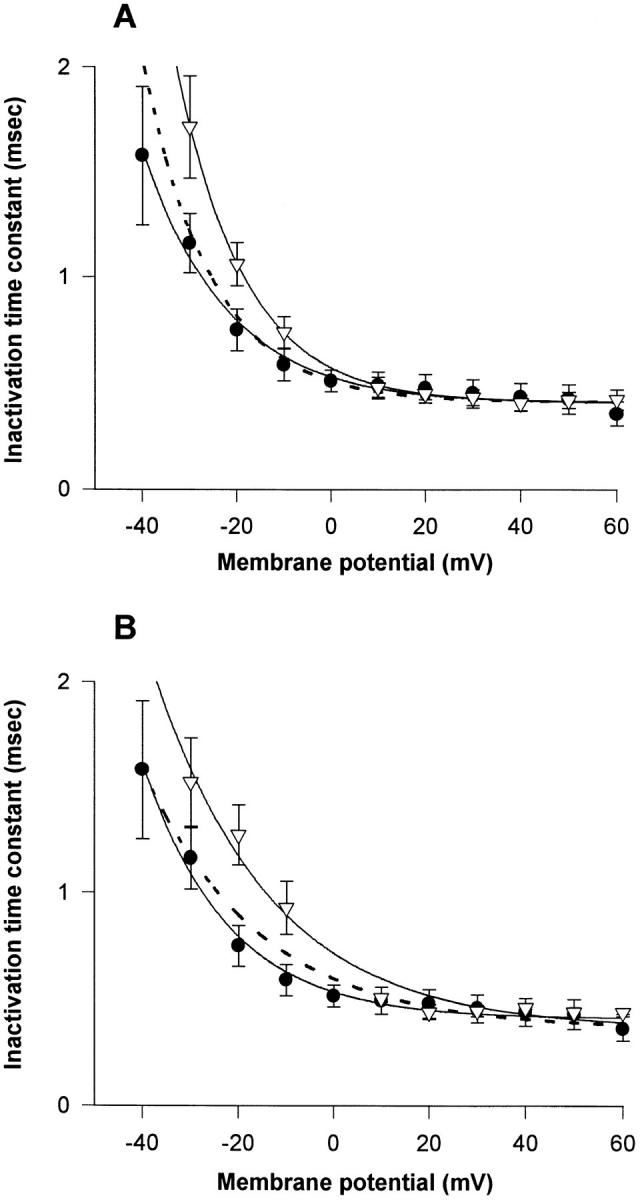
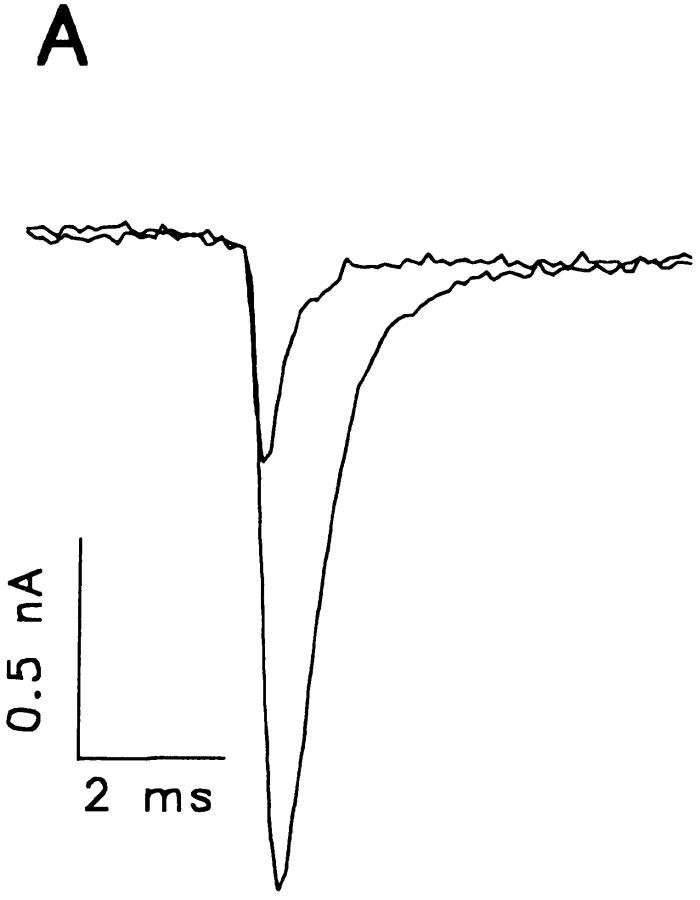
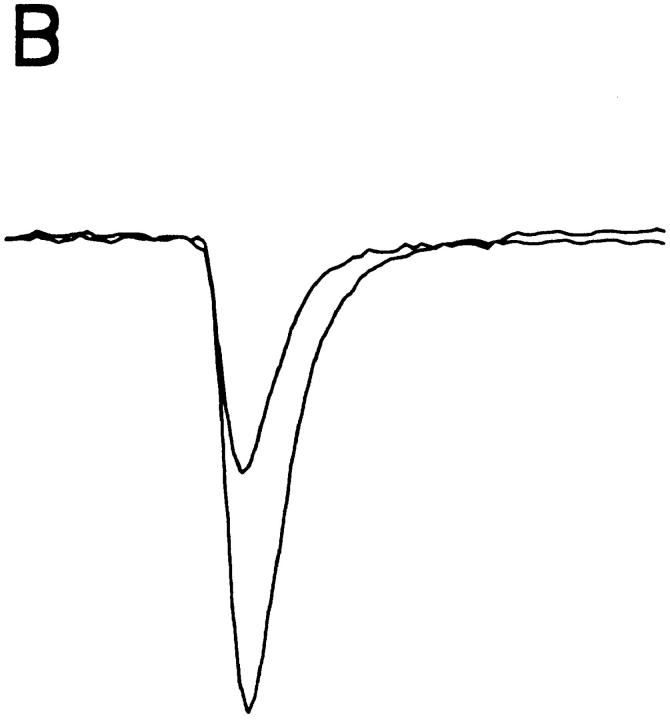
A potential role for sialic acid in the voltage-dependent gating of rat skeletal muscle sodium channels (rSkM1) was investigated using Chinese hamster ovary (CHO) cells stably transfected with rSkM1. Changes in the voltage dependence of channel gating were observed after enzymatic (neuraminidase) removal of sialic acid from cells expressing rSkM1 and through the expression of rSkM1 in a sialylation-deficient cell line (lec2). The steady-state half-activation voltages (Va) of channels under each condition of reduced sialylation were approximately 10 mV more depolarized than control channels. The voltage dependence of the time constants of channel activation and inactivation were also shifted in the same direction and by a similar magnitude. In addition, recombinant deletion of likely glycosylation sites from the rSkM1 sequence resulted in mutant channels that gated at voltages up to 10mV more positive than wild-type channels. Thus three independent means of reducing channel sialylation show very similar effects on the voltage dependence of channel gating. Finally, steady-state activation voltages for channels subjected to reduced sialylation conditions were much less sensitive to the effects of external calcium than those measured under control conditions, indicating that sialic acid directly contributes to the negative surface potential. These results are consistent with an electrostatic mechanism by which external, negatively charged sialic acid residues on rSkM1 alter the electric field sensed by channel gating elements.
Figures





References
-
- Chen CA, Okayama H. Calcium-phosphate-mediated gene transfer: A highly efficient transfection system for stably transforming cells with plasmid DNA. Biotechniques. 1988;6:632–638. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
