

Nitric oxide is the mediator of both endothelium-dependent relaxation and hyperpolarization of the rabbit carotid artery
- PMID: 9108128
- PMCID: PMC20600
- DOI: 10.1073/pnas.94.8.4193
Nitric oxide is the mediator of both endothelium-dependent relaxation and hyperpolarization of the rabbit carotid artery
Abstract
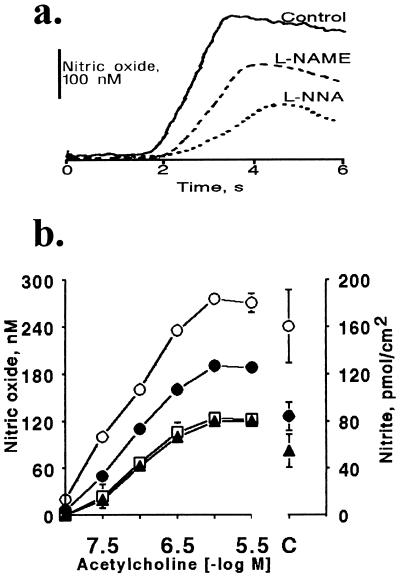
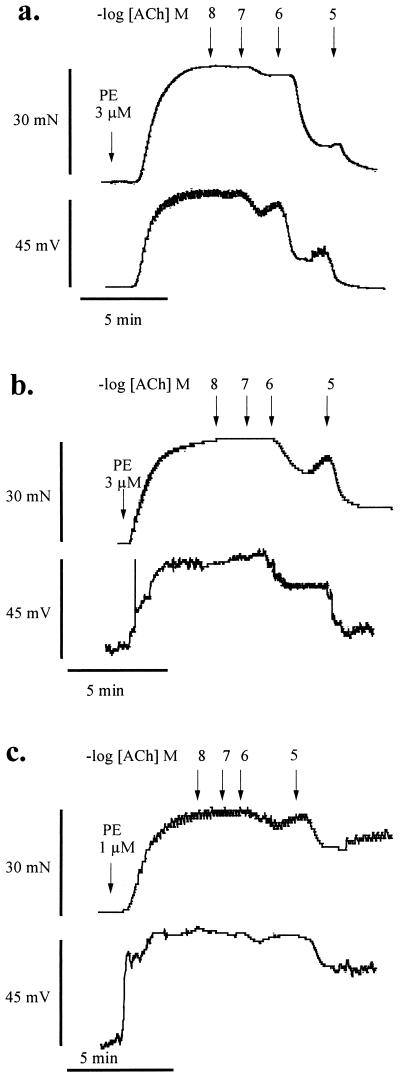
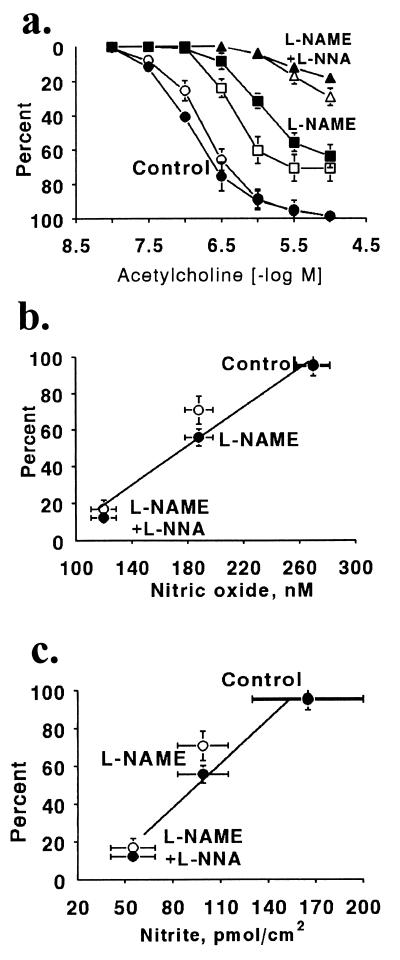
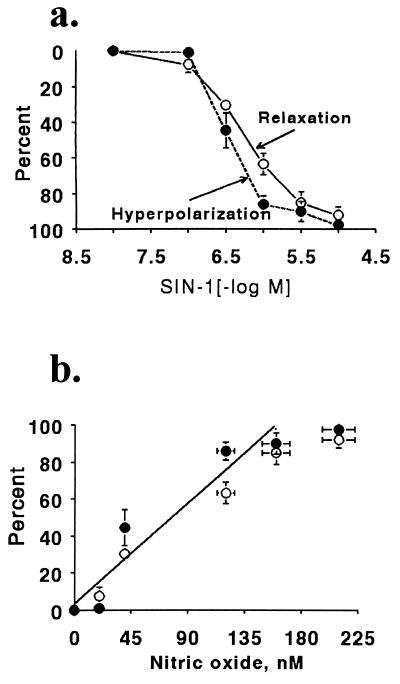
It is controversial whether the endothelial cell release of nitric oxide (NO) or a different factor(s) accounts for endothelium-dependent hyperpolarization, because in many arteries endothelium-dependent relaxation and hyperpolarization resists inhibitors of NO synthase. The contribution of NO to acetylcholine-induced endothelium-dependent hyperpolarization and relaxation of the rabbit carotid artery was determined by measuring NO with electrochemical and chemiluminescence techniques. In the presence of phenylephrine to depolarize and contract the smooth muscle cells, acetylcholine caused concentration-dependent hyperpolarization and relaxation which were closely correlated to the release of NO. N(omega)-nitro-L-arginine methyl ester (30 microM) partially reduced the release of NO and caused a similar reduction in smooth muscle cell relaxation and hyperpolarization. To determine if the residual responses were mediated by another endothelium-derived mediator or NO released despite treatment with N(omega)-nitro-L-arginine methyl ester, N(omega)-nitro-L-arginine (300 microM) was added. The combined inhibitors further reduced, but did not eliminate, NO release, smooth muscle relaxation, and hyperpolarization. Hyperpolarization and relaxation to acetylcholine remained closely correlated with the release of NO in the presence of the inhibitors. In addition, the NO donor, SIN-1, caused hyperpolarization and relaxation which correlated with the concentrations of NO that it released. These studies indicate that (i) the release of NO by acetylcholine is only partially inhibited by these inhibitors of NO synthase when used even at high concentrations, and (ii) NO rather than another factor accounts fully for endothelium-dependent responses of the rabbit carotid artery.
Figures
References
-
- Feelisch M, te Poel M, Zamora R, Deussen A, Moncada S. Nature (London) 1994;368:62–65. - PubMed
-
- Cohen R A, Vanhoutte P M. Circulation. 1995;92:3337–3349. - PubMed
-
- Garland C J, Plane F, Kemp B K, Cocks T M. Trends Pharmacol Sci. 1995;16:23–30. - PubMed
-
- Cowan C L, Cohen R A. Am J Physiol. 1991;261:H830–H835. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
