

Mechanisms and effects of intracellular calcium buffering on neuronal survival in organotypic hippocampal cultures exposed to anoxia/aglycemia or to excitotoxins
- PMID: 9133378
- PMCID: PMC6573708
- DOI: 10.1523/JNEUROSCI.17-10-03538.1997
Mechanisms and effects of intracellular calcium buffering on neuronal survival in organotypic hippocampal cultures exposed to anoxia/aglycemia or to excitotoxins
Abstract
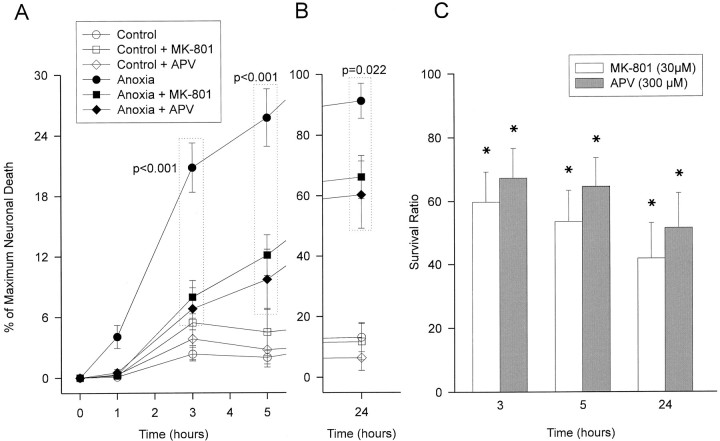
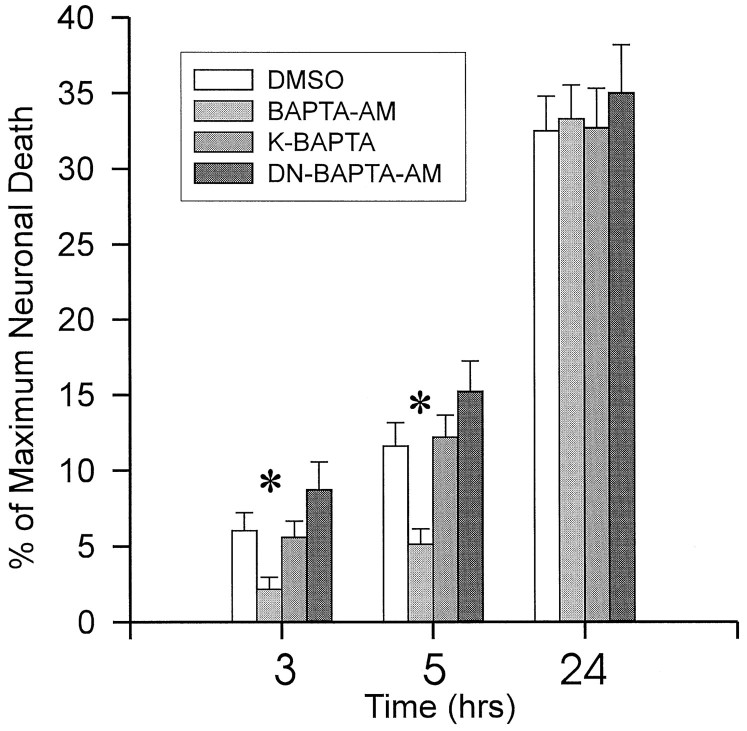
Neuronal calcium loading attributable to hypoxic/ischemic injury is believed to trigger neurotoxicity. We examined in organotypic hippocampal slice cultures whether artificially and reversibly enhancing the Ca2+ buffering capacity of neurons reduces the neurotoxic sequelae of oxygen-glucose deprivation (OGD), whether such manipulation has neurotoxic potential, and whether the mechanism underlying these effects is pre- or postsynaptic. Neurodegeneration caused over 24 hr by 60 min of OGD was triggered largely by NMDA receptor activation and was attenuated temporarily by pretreating the slices with cell-permeant Ca2+ buffers such as 1, 2 bis(2-aminophenoxy)ethane-N,N,N',N'-tetra-acetic acid acetoxymethyl ester (BAPTA-AM). This pretreatment produced a transient, reversible increase in intracellular buffer content as demonstrated autoradiographically using slices loaded with 14C-BAPTA-AM and by confocal imaging of slices loaded with the BAPTA-AM analog calcium green-acetoxymethyl ester (AM). The time courses of 14C-BAPTA retention and of neuronal survival after OGD were identical, indicating that increased buffer content is necessary for the observed protective effect. Protection by Ca2+ buffering originated presynaptically because BAPTA-AM was ineffective when endogenous transmitter release was bypassed by directly applying NMDA to the cultures, and because pretreatment with the low Ca2+ affinity buffer 2-aminophenol-N,N,O-triacetic acid acetoxymethyl ester, which attenuates excitatory transmitter release, attenuated neurodegeneration. Thus, in cultured hippocampal slices, enhancing neuronal Ca2+ buffering unequivocally attenuates or delays the onset of anoxic neurodegeneration, likely by attenuating the synaptic release of endogenous excitatory neurotransmitters (excitotoxicity).
Figures








References
-
- Abdel-Hamid KM (1994) The regulation of intracellular calcium in cultured hippocampal neurons and the influence of calcium buffers. PhD thesis, University of British Columbia.
-
- Arkhammar P, Nilsson T, Bergmann PO. Glucose mediated efflux of indo-1 from pancreatic B-cells is reduced by probenecid. FEBS Lett. 1991;273:182–184. - PubMed
-
- Baimbridge KG, Abdel-Hamid KM. Intra-neuronal Ca2+-buffering with BAPTA enhances glutamate excitotoxicity in vitro and ischemic damage in vivo. Soc Neurosci Abstr. 1992;18:571.4.
-
- Bigelow DJ, Squier TC, Thomas DD. Temperature dependence of rotational dynamics of protein and lipid in sarcoplasmic reticulum membranes. Biochemistry. 1986;25:194–202. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous