

Cross-validated maximum likelihood enhances crystallographic simulated annealing refinement
- PMID: 9144182
- PMCID: PMC24623
- DOI: 10.1073/pnas.94.10.5018
Cross-validated maximum likelihood enhances crystallographic simulated annealing refinement
Abstract
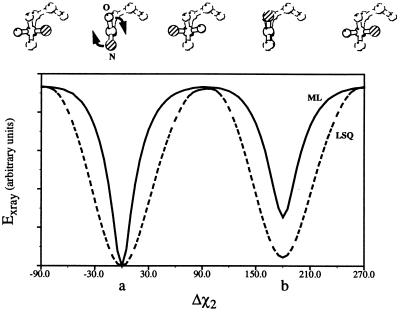
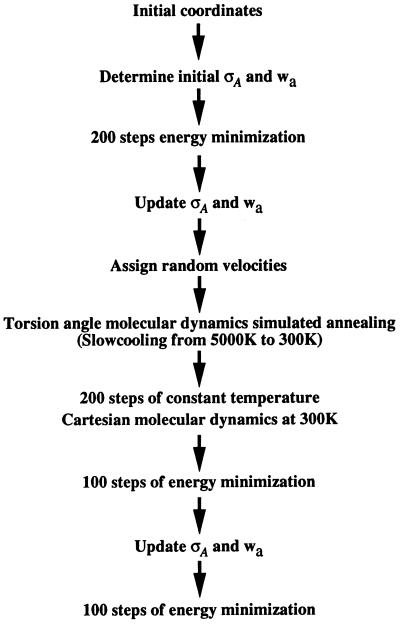
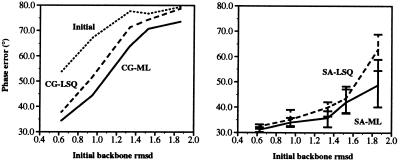
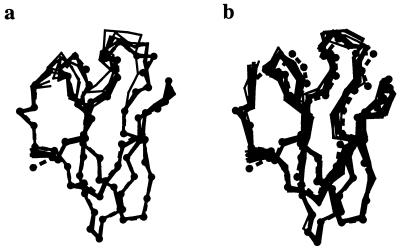
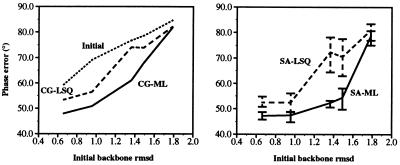
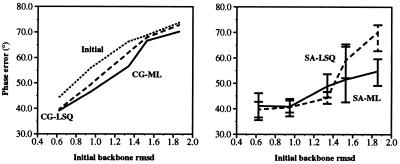
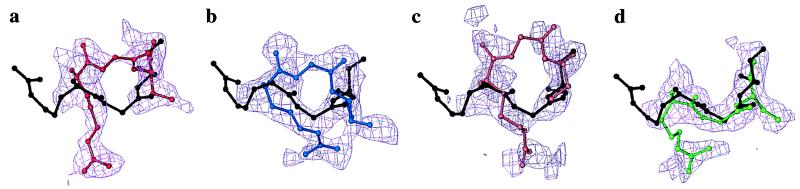
Recently, the target function for crystallographic refinement has been improved through a maximum likelihood analysis, which makes proper allowance for the effects of data quality, model errors, and incompleteness. The maximum likelihood target reduces the significance of false local minima during the refinement process, but it does not completely eliminate them, necessitating the use of stochastic optimization methods such as simulated annealing for poor initial models. It is shown that the combination of maximum likelihood with cross-validation, which reduces overfitting, and simulated annealing by torsion angle molecular dynamics, which simplifies the conformational search problem, results in a major improvement of the radius of convergence of refinement and the accuracy of the refined structure. Torsion angle molecular dynamics and the maximum likelihood target function interact synergistically, the combination of both methods being significantly more powerful than each method individually. This is demonstrated in realistic test cases at two typical minimum Bragg spacings (dmin = 2.0 and 2.8 A, respectively), illustrating the broad applicability of the combined method. In an application to the refinement of a new crystal structure, the combined method automatically corrected a mistraced loop in a poor initial model, moving the backbone by 4 A.
Figures
References
-
- Hendrickson W A. Methods Enzymol. 1985;115:252–270. - PubMed
-
- Tronrud D E, Ten Eyck L F, Matthews B W. Acta Crystallogr A. 1987;43:489–500.
-
- Jack A, Levitt M. Acta Crystallogr A. 1978;34:931–935.
-
- Brünger A T, Kuriyan J, Karplus M. Science. 1987;235:458–460. - PubMed
-
- Gros P, Betzel C, Dauter Z, Wilson K S, Hol W G J. J Mol Biol. 1989;210:347–367. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
