

Elevation of intracellular calcium in smooth muscle causes endothelial cell generation of NO in arterioles
- PMID: 9177252
- PMCID: PMC21084
- DOI: 10.1073/pnas.94.12.6529
Elevation of intracellular calcium in smooth muscle causes endothelial cell generation of NO in arterioles
Abstract
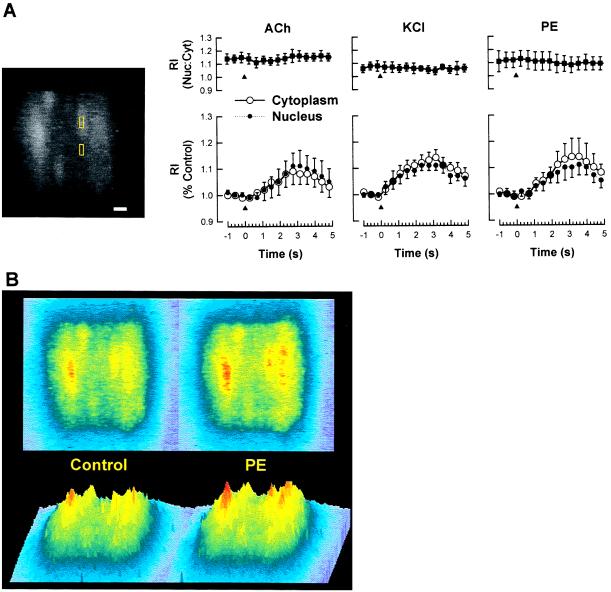
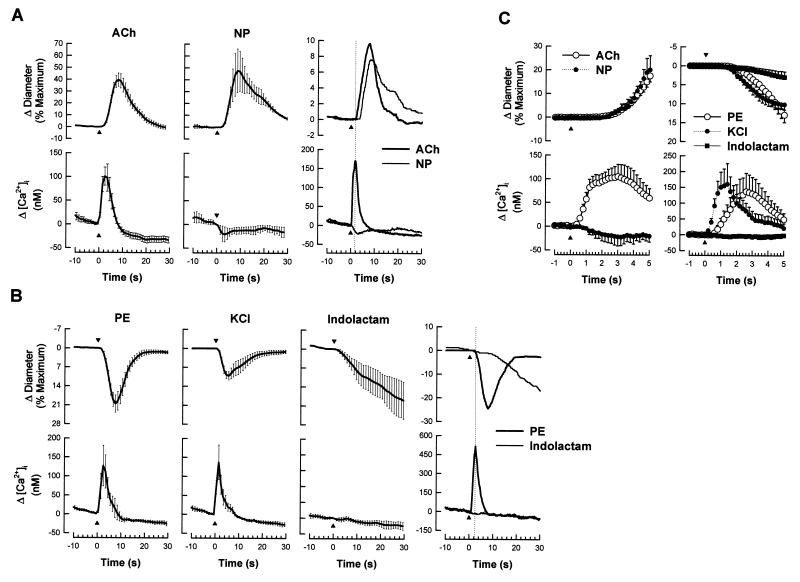
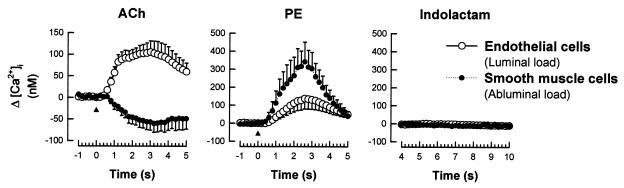
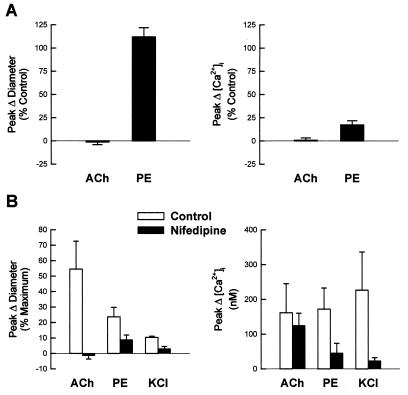
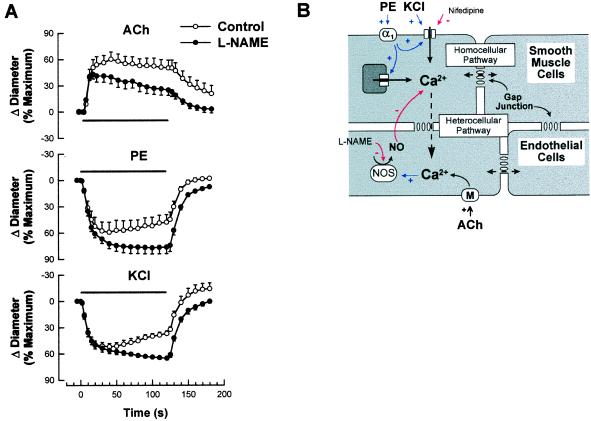
It is well known that vascular smooth muscle tone can be modulated by signals arising in the endothelium (e.g., endothelium-derived relaxing factor, endothelium-derived hyperpolarizing factor, and prostaglandins). Here we show that during vasoconstriction a signal can originate in smooth muscle cells and act on the endothelium to cause synthesis of endothelium-derived relaxing factor. We studied responses to two vasoconstrictors (phenylephrine and KCl) that act by initiating a rise in smooth muscle cell intracellular Ca2+ concentration ([Ca2+]i) and exert little or no direct effect on the endothelium. Fluo-3 was used as a Ca2+ indicator in either smooth muscle or endothelial cells of arterioles from the hamster cheek pouch. Phenylephrine and KCl caused the expected rise in smooth muscle cell [Ca2+]i that was accompanied by an elevation in endothelial cell [Ca2+]i. The rise in endothelial cell [Ca2+]i was followed by increased synthesis of NO, as evidenced by an enhancement of the vasoconstriction induced by both agents after blockade of NO synthesis. The molecule involved in signal transmission from smooth muscle to endothelium is as yet unknown. However, given that myoendothelial cell junctions are frequent in these vessels, we hypothesize that the rise in smooth muscle cell Ca2+ generates a diffusion gradient that drives Ca2+ through myoendothelial cell junctions and into the endothelial cells, thereby initiating the synthesis of NO.
Figures
References
-
- Busse, R., Pohl, U. & Luckhoff, A. (1989) Z. Kardiol. 78, Suppl. 6, 64–69. - PubMed
-
- von der Weid P Y, Beny J L. Am J Physiol. 1992;262:H1823–H1831. - PubMed
-
- Angus J A, Cocks T M, Satoh K. Fed Proc Fed Am Soc Exp Biol. 1986;45:2355–2359. - PubMed
-
- Amerini S, Mantelli L, Ledda F. Pharmacol Res. 1995;31:175–181. - PubMed
-
- Ayajiki K, Kindermann M, Hecker M, Fleming I, Busse R. Circ Res. 1996;78:750–758. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
