

PowerBLAST: a new network BLAST application for interactive or automated sequence analysis and annotation
- PMID: 9199938
- PMCID: PMC310664
- DOI: 10.1101/gr.7.6.649
PowerBLAST: a new network BLAST application for interactive or automated sequence analysis and annotation
Abstract
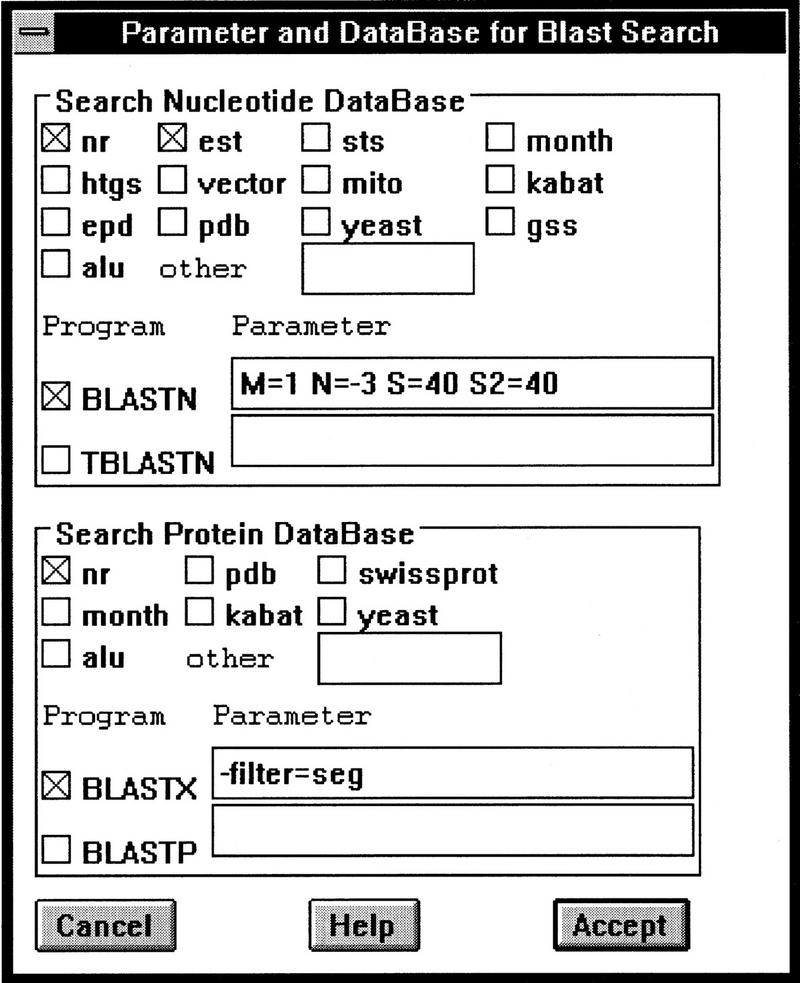
As the rate of DNA sequencing increases, analysis by sequence similarity search will need to become much more efficient in terms of sensitivity, specificity, automation potential, and consistency in annotation. PowerBLAST was developed, in part, to address these problems. PowerBLAST includes a number of options for masking repetitive elements and low complexity subsequences. It also has the capacity to restrict the search to any level of NCBI's taxonomy index, thus supporting "comparative genomics" applications. Postprocessing of the BLAST output using the SIM series of algorithms produces optimal, gapped alignments, and multiple alignments when a region of the query sequence matches multiple database sequences. PowerBLAST is capable of processing sequences of any length because it divides long query sequences into overlapping fragments and then merges the results after searching. The results may be viewed graphically, as a textual representation, or as an HTML page with links to GenBank and Entrez. For matching database sequences, annotated features are superimposed on the aligned query sequence in the output, thus greatly increasing the ease of interpretation. Such features may be used for automated annotation of new sequence because PowerBLAST output in ASN.1 form may be "dragged and dropped" into NCBI's Sequin program for sequence annotation and submission. PowerBLAST is capable of analyzing and annotating a 100-kb query in 60 min on NCBI's BLAST server.
Figures
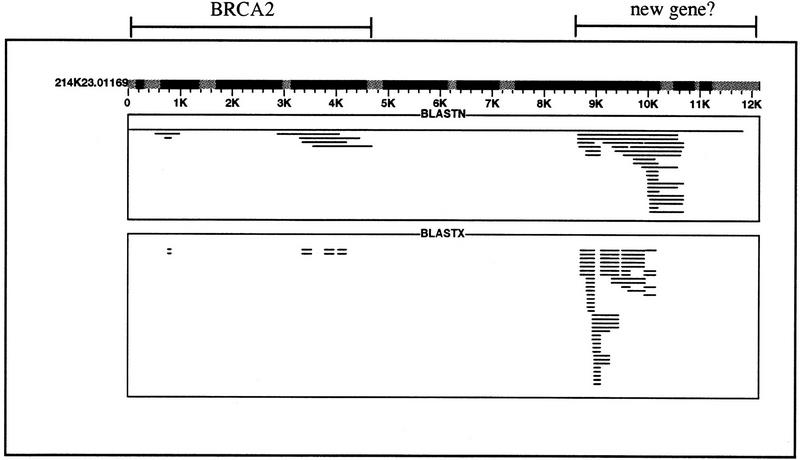
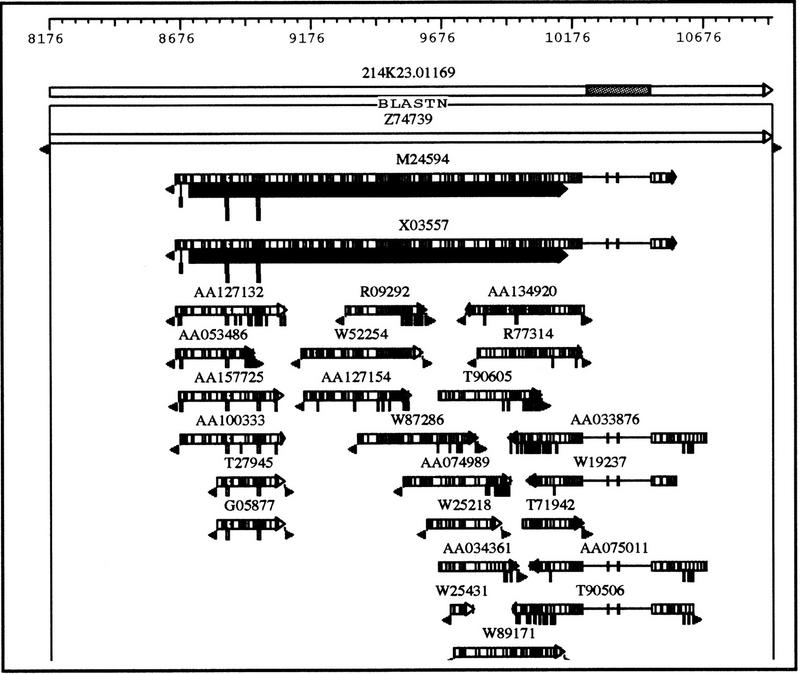
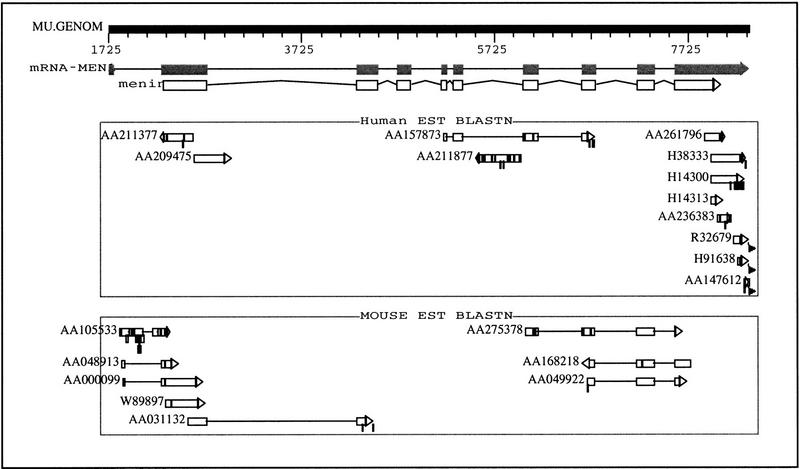

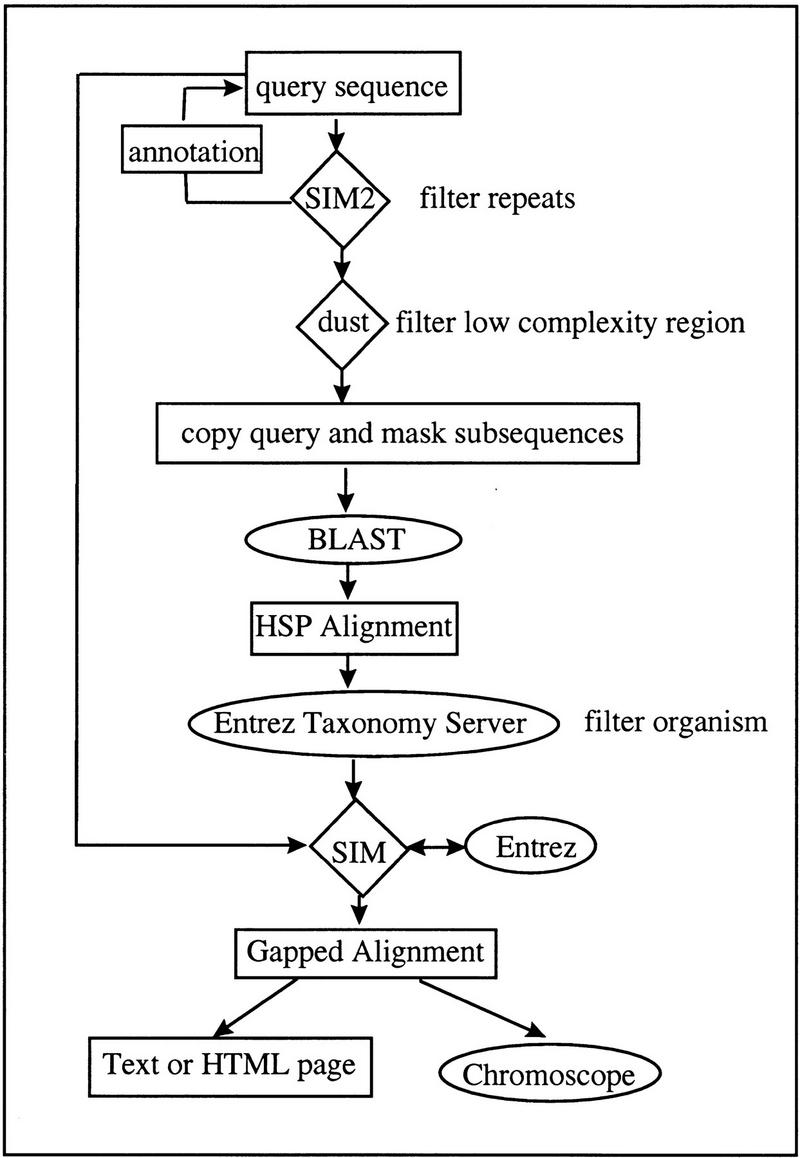
References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
-
- Altschul SF, Boguski MS, Wootton JC. Issues in searching molecular sequence databases. Nature Genet. 1994;6:119–129. - PubMed
-
- Chao K-M, Zhang J, Ostell J, Miller W. A local alignment tool for very long DNA sequences. Comput Applic Biosci. 1994;11:147–153. - PubMed
-
- Chao K-M, Zhang J, Ostell J, Miller W. A tool for aligning very similar DNA sequences. Comput Applic Biosci. 1997;13:75–80. - PubMed
-
- Chandrasekharappa SC, Guru SC, Manickam P, Olufemi S, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, Crabtree JS, Wang Y, Roe BA, Weisemann J, Boguski MS, Agarwal SK, Kester MB, Kim YS, Heppner C, Dong Q, Spiegel AM, Burns AL, Marx SJ. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–407. - PubMed
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials