

Transgenic Galphaq overexpression induces cardiac contractile failure in mice
- PMID: 9223325
- PMCID: PMC21567
- DOI: 10.1073/pnas.94.15.8121
Transgenic Galphaq overexpression induces cardiac contractile failure in mice
Abstract
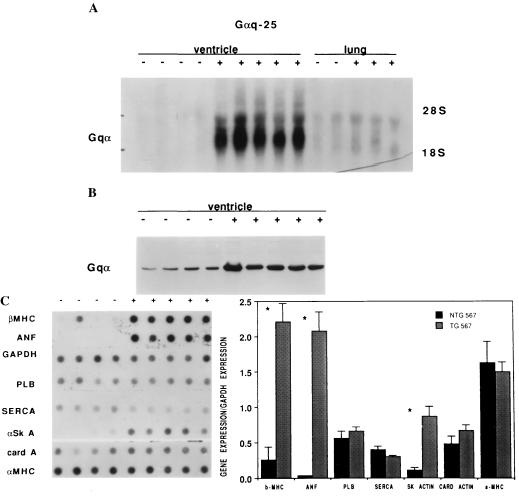
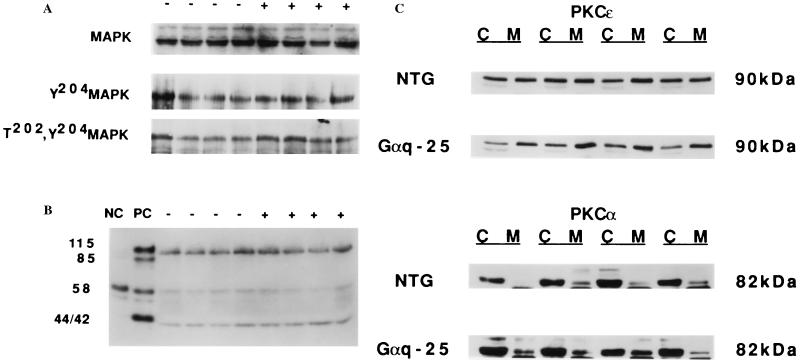
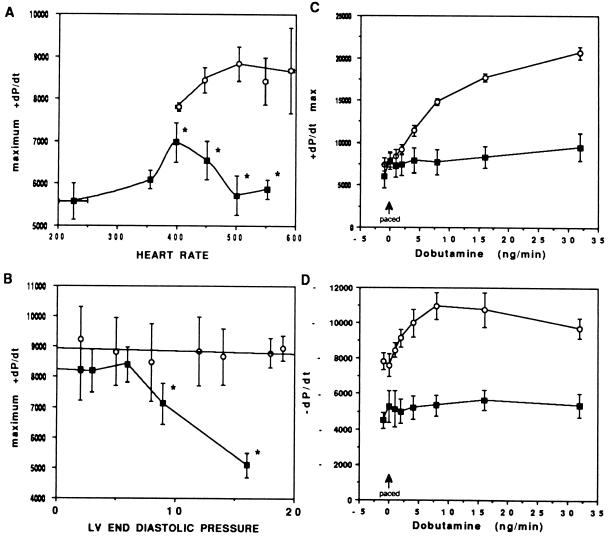
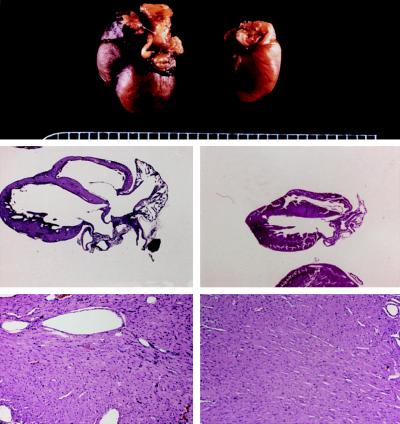
The critical cell signals that trigger cardiac hypertrophy and regulate the transition to heart failure are not known. To determine the role of Galphaq-mediated signaling pathways in these events, transgenic mice were constructed that overexpressed wild-type Galphaq in the heart using the alpha-myosin heavy chain promoter. Two-fold overexpression of Galphaq showed no detectable effects, whereas 4-fold overexpression resulted in increased heart weight and myocyte size along with marked increases in atrial naturietic factor ( approximately 55-fold), beta-myosin heavy chain ( approximately 8-fold), and alpha-skeletal actin ( approximately 8-fold) expression, and decreased ( approximately 3-fold) beta-adrenergic receptor-stimulated adenylyl cyclase activity. All of these signals have been considered markers of hypertrophy or failure in other experimental systems or human heart failure. Echocardiography and in vivo cardiac hemodynamic studies indeed revealed impaired intrinsic contractility manifested as decreased fractional shortening (19 +/- 2% vs. 41 +/- 3%), dP/dt max, a negative force-frequency response, an altered Starling relationship, and blunted contractile responses to the beta-adrenergic agonist dobutamine. At higher levels of Galphaq overexpression, frank cardiac decompensation occurred in 3 of 6 animals with development of biventricular failure, pulmonary congestion, and death. The element within the pathway that appeared to be critical for these events was activation of protein kinase Cepsilon. Interestingly, mitogen-activated protein kinase, which is postulated by some to be important in the hypertrophy program, was not activated. The Galphaq overexpressor exhibits a biochemical and physiologic phenotype resembling both the compensated and decompensated phases of human cardiac hypertrophy and suggests a common mechanism for their pathogenesis.
Figures
References
-
- Shubeita H E, McDonough P M, Harris A N, Knowlton K U, Glembotski C C, Brown J H, Chien K R. J Biol Chem. 1990;265:20555–20562. - PubMed
-
- Sadoshima J-I, Xu Y, Slayter H S, Izumo S. Cell. 1993;75:977–984. - PubMed
-
- Knowlton K U, Michel M C, Itani M, Shubeita H E, Ishihara K, Brown J H, Chien K R. J Biol Chem. 1993;268:15374–15380. - PubMed
-
- Adams J W, Migita D S, Yu M K, Young R, Hellickson M S, Castro-Vargas F E, Domingo J D, Lee P H, Bui J S, Henderson S A. J Biol Chem. 1996;271:1179–1186. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
