

The hepatitis B virus X gene induces p53-mediated programmed cell death
- PMID: 9223332
- PMCID: PMC21574
- DOI: 10.1073/pnas.94.15.8162
The hepatitis B virus X gene induces p53-mediated programmed cell death
Abstract
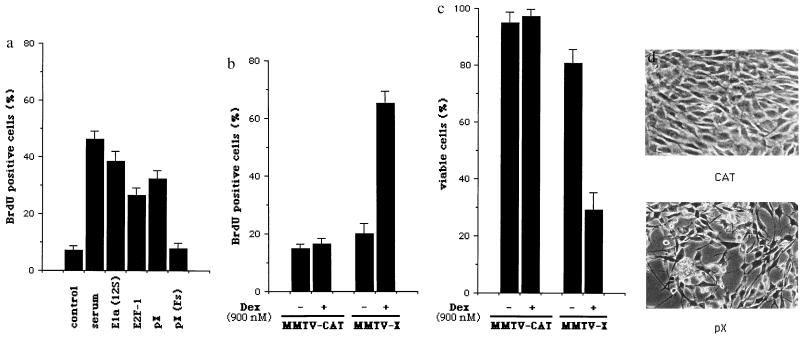
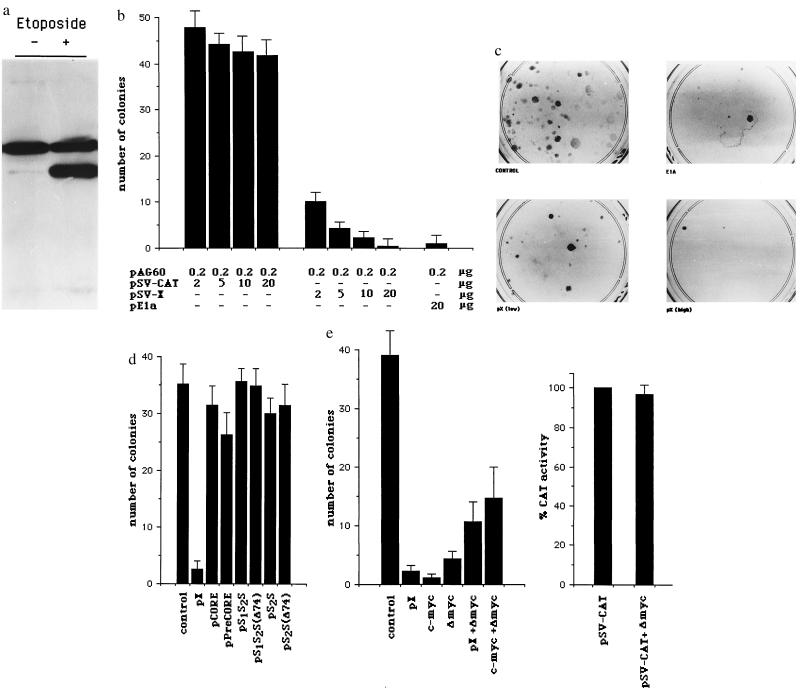
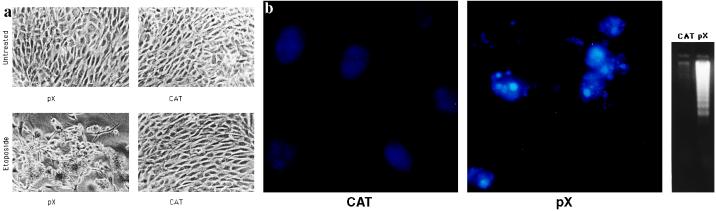
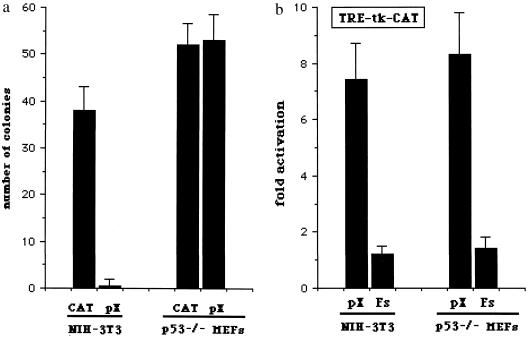
The human hepatitis B virus (HBV) protein pX is a multifunctional regulatory protein that is known to affect both transcription and cell growth. Here we describe induction of apoptosis in NIH 3T3 polyclonal cell lines upon stimulation of pX expression from a dexamethasone inducible mouse mammary tumor virus (MMTV)-X expression vector. The effect of long-term pX expression on the cell survival of mouse fibroblasts was confirmed in colony generation assays. This effect is not shared either by the other HBV products and it is c-myc mediated, as shown by the use of a dominant negative deletion mutant of c-myc. pX also sensitize cells to programmed cell death after exposure to DNA damaging agents. Taking advantage of stable transfectants carrying the p53val135 temperature-sensitive allele, we directly demonstrate that induction of apoptosis by pX requires p53. In p53 null mouse embryo fibroblasts pX activates transcription and confers an evident growth advantage without loss of cell viability. Although pX protein was not detectable in the experimental conditions we used, our results indicate that its expression affects both cell growth and cell death control.
Figures
References
-
- Avantaggiati M L, Natoli G, Balsano C, Chirillo P, Artini M, De Marzio E, Collepardo D, Levrero M. Oncogene. 1993;8:1567–1574. - PubMed
-
- Twu J S, Lai M Y, Chen D S, Robinson W S. Virology. 1993;192:346–350. - PubMed
-
- Balsano C, Avantaggiati M L, Natoli G, De Marzio E, Will H, Perricaudet M, Levrero M. Biochem Biophys Res Commun. 1991;176:985–992. - PubMed
-
- Menzo S, Clementi M, Alfani E, Bagnarelli P, Iacovacci S, Manzin A, Dandri M, Natoli G, Levrero M, Carloni G. Virology. 1993;196:878–882. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
