

Plasma membrane translocation of fluorescent-labeled phosphatidylethanolamine is controlled by transcription regulators, PDR1 and PDR3
- PMID: 9230069
- PMCID: PMC2138184
- DOI: 10.1083/jcb.138.2.255
Plasma membrane translocation of fluorescent-labeled phosphatidylethanolamine is controlled by transcription regulators, PDR1 and PDR3
Abstract
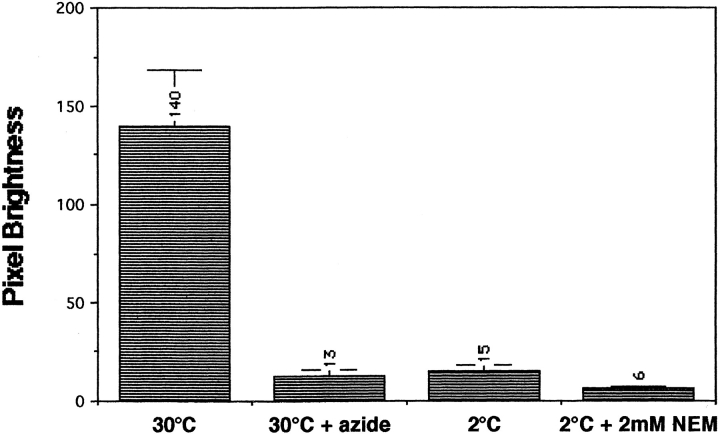
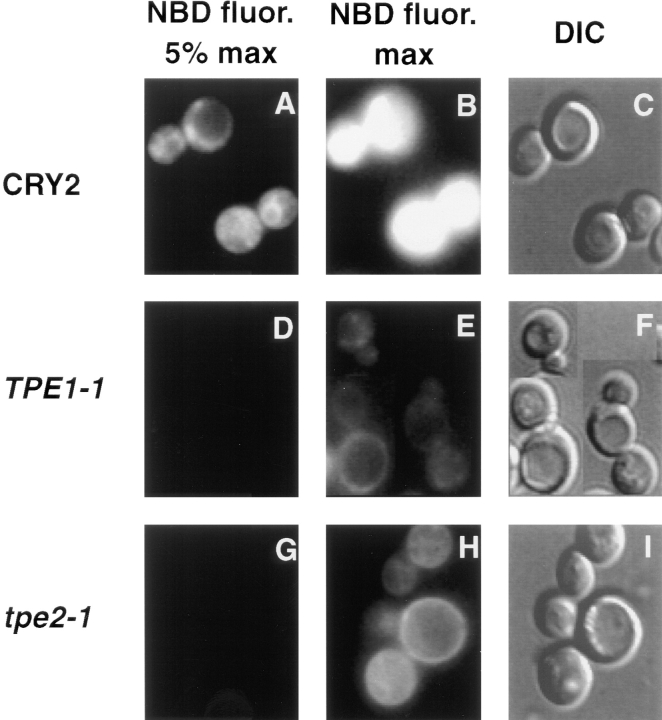
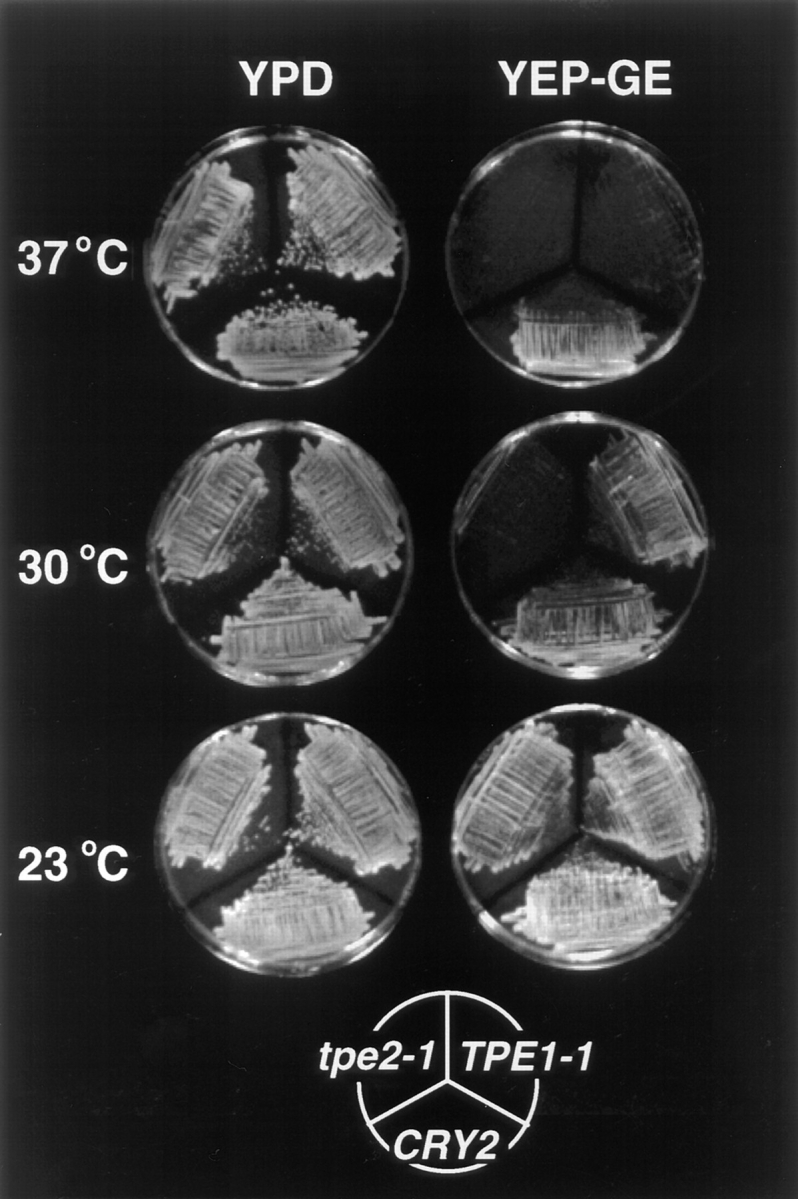
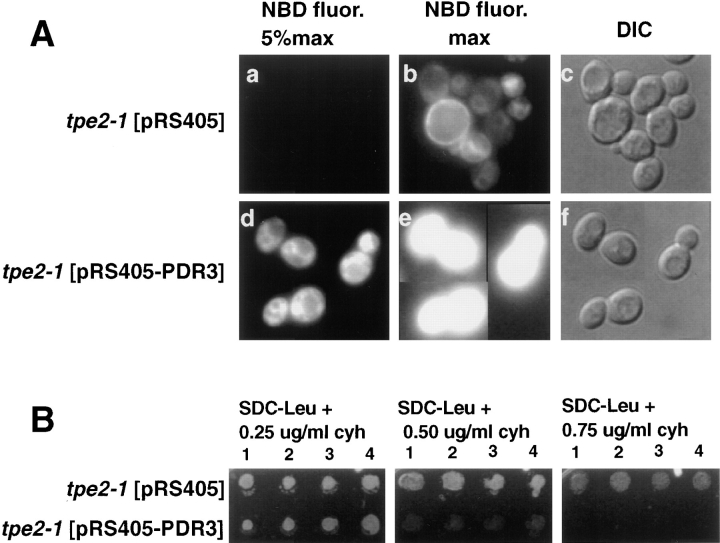
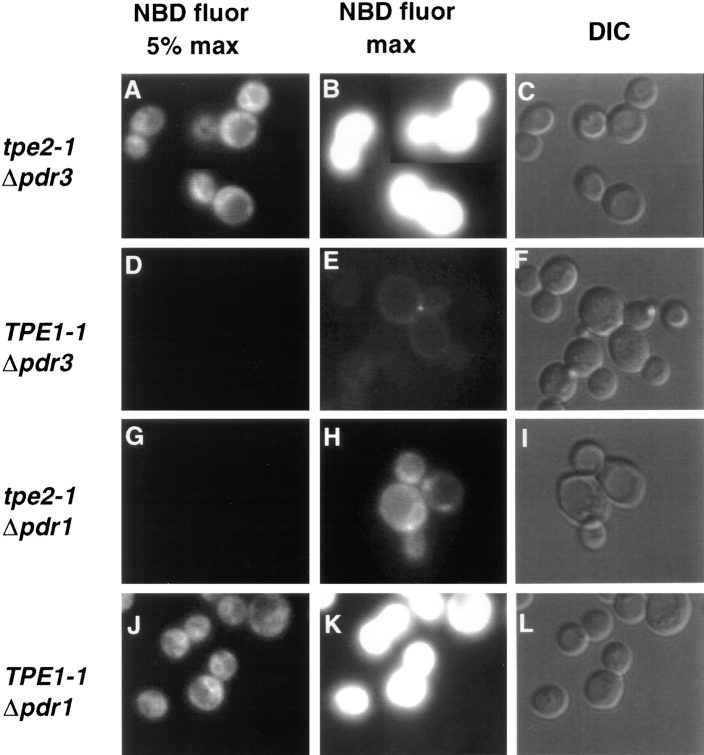
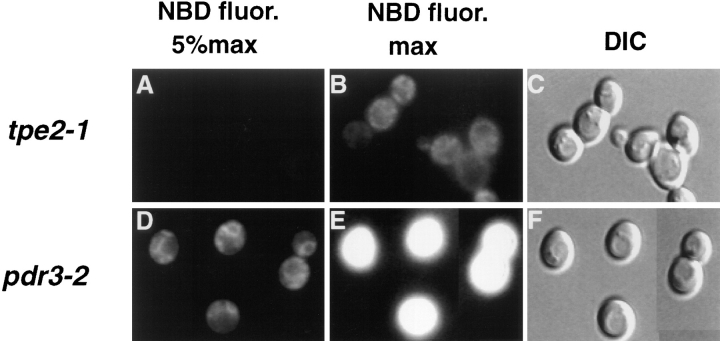
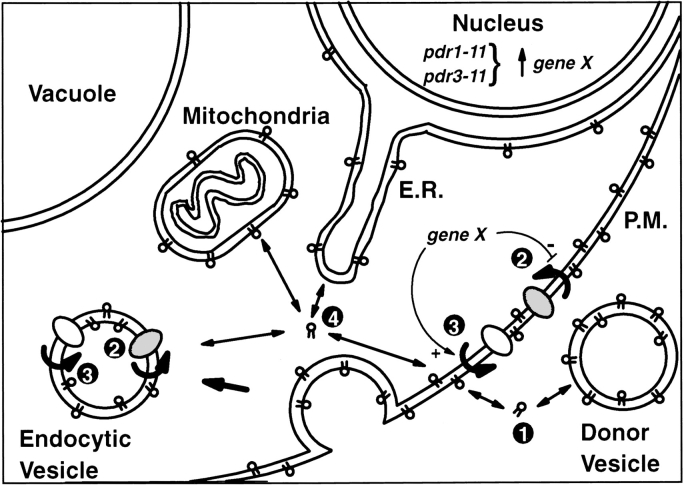
The transcription regulators, PDR1 and PDR3, have been shown to activate the transcription of numerous genes involved in a wide range of functions, including resistance to physical and chemical stress, membrane transport, and organelle function in Saccharomyces cerevisiae. We report here that PDR1 and PDR3 also regulate the transcription of one or more undetermined genes that translocate endogenous and fluorescent-labeled (M-C6-NBD-PE) phosphatidylethanolamine across the plasma membrane. A combination of fluorescence microscopy, fluorometry, and quantitative analysis demonstrated that M-C6-NBD-PE can be translocated both inward and outward across the plasma membrane of yeast cells. Mutants, defective in the accumulation of M-C6-NBD-PE, were isolated by selectively photokilling normal cells that accumulated the fluorescent phospholipid. This led to the isolation of numerous trafficking in phosphatidylethanolamine (tpe) mutants that were defective in intracellular accumulation of M-C6-NBD-PE. Complementation cloning and linkage analysis led to the identification of the dominant mutation TPE1-1 as a new allele of PDR1 and the semidominant mutation tpe2-1 as a new allele of PDR3. The amount of endogenous phosphatidylethanolamine exposed to the outer leaflet of the plasma membrane was measured by covalent labeling with the impermeant amino reagent, trinitrobenzenesulfonic acid. The amount of outer leaflet phosphatidylethanolamine in both mutant strains increased four- to fivefold relative to the parent Tpe+ strain, indicating that the net inward flux of endogenous phosphatidylethanolamine as well as M-C6-NBD-PE was decreased. Targeted deletions of PDR1 in the new allele, PDR1-11, and PDR3 in the new allele, pdr3-11, resulted in normal M-C6-NBD-PE accumulation, confirming that PDR1-11 and pdr3-11 were gain-of-function mutations in PDR1 and PDR3, respectively. Both mutant alleles resulted in resistance to the drugs cycloheximide, oligomycin, and 4-nitroquinoline N-oxide (4-NQO). However, a previously identified drug-resistant allele, pdr3-2, accumulated normal amounts of M-C6-NBD-PE, indicating allele specificity for the loss of M-C6-NBD-PE accumulation. These data demonstrated that PDR1 and PDR3 regulate the net rate of M-C6-NBD-PE translocation (flip-flop) and the steady-state distribution of endogenous phosphatidylethanolamine across the plasma membrane.
Figures
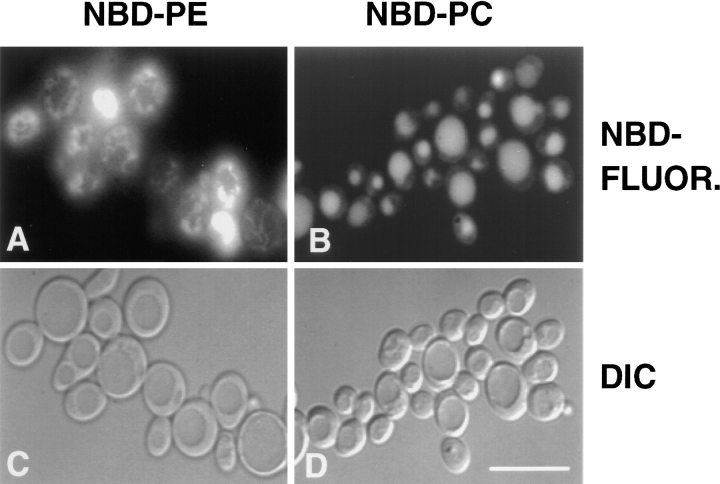
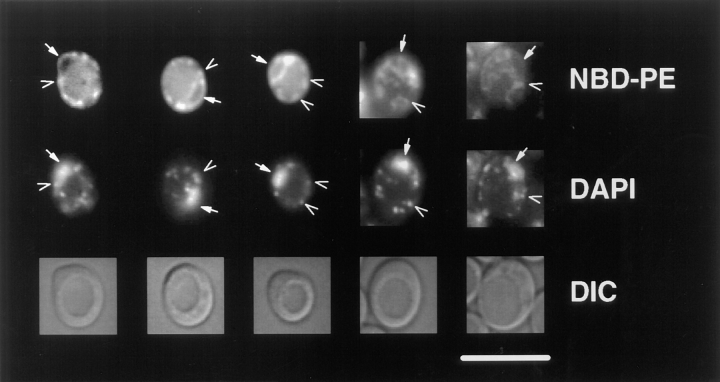
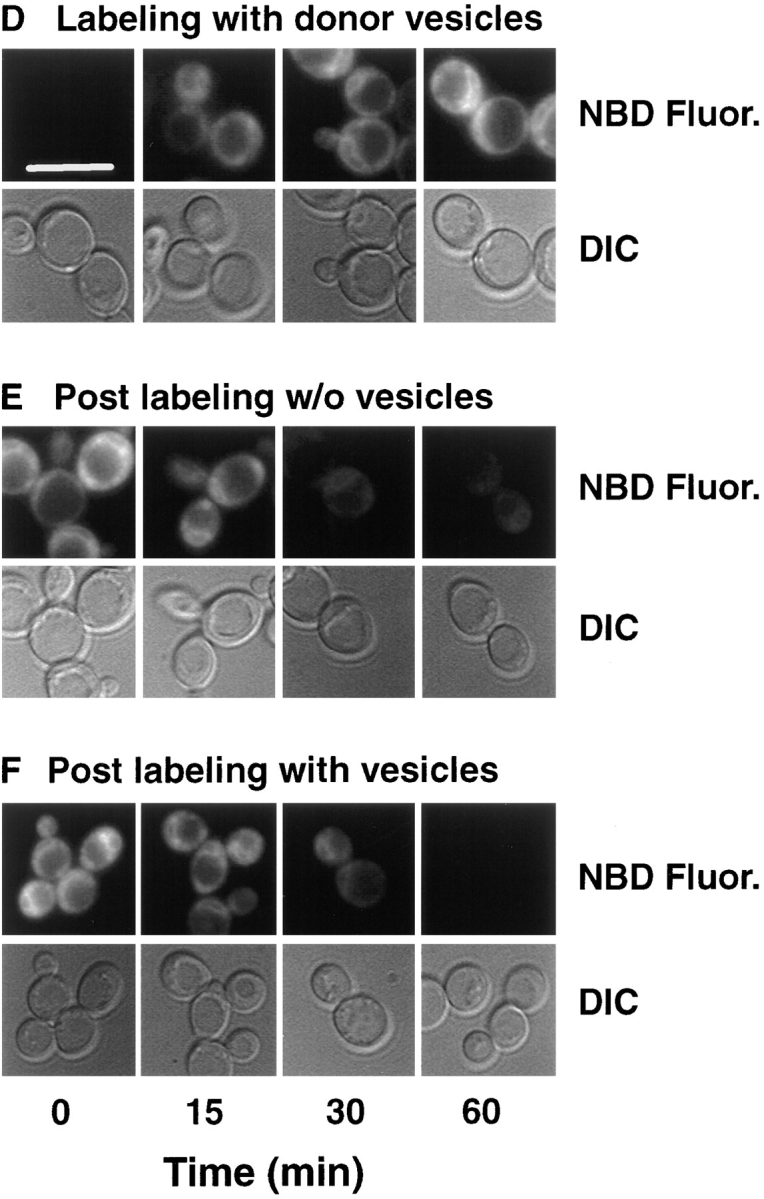
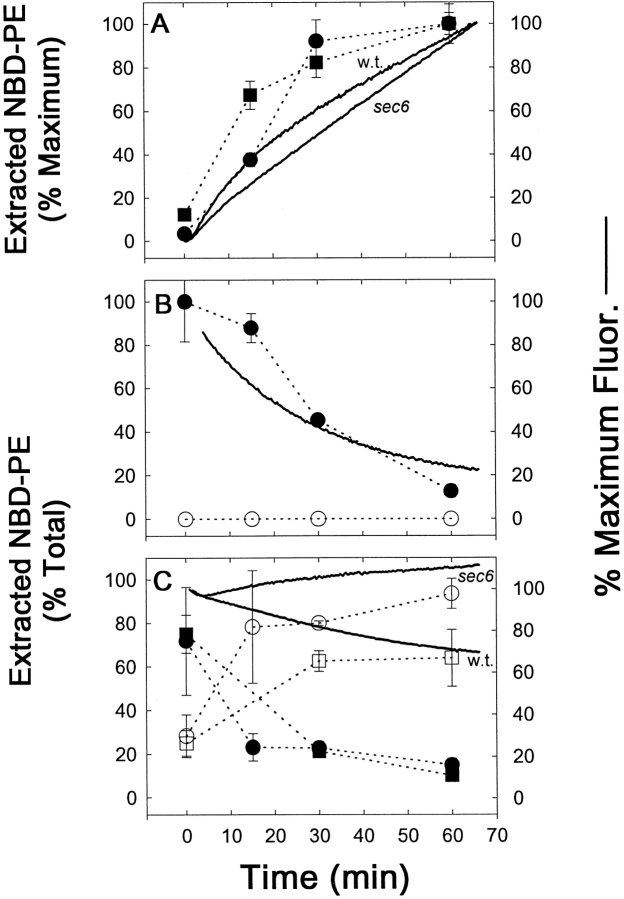
References
-
- Ames BN, Dubin DT. The role of polyamines in the neutralization of bacteriophage deoxyribonucleic acid. J Biol Chem. 1960;235:769–775. - PubMed
-
- Balasubramanian K, Gupta CM. Transbilayer phosphatidylethanolamine movements in the yeast plasma membrane. Evidence for a protein-mediated, energy-dependent mechanism. Eur J Biochem. 1996;240:798–806. - PubMed
-
- Balzi E, Goffeau A. Genetics and biochemistry of yeast multidrug resistance. Biochim Biophys Acta. 1994;1187:152–162. - PubMed
-
- Balzi E, Goffeau A. Yeast multidrug resistance: the PDR network. J Bioenerg Biomembr. 1995;27:71–76. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
