

CD36 mediates the In vitro inhibitory effects of thrombospondin-1 on endothelial cells
- PMID: 9245797
- PMCID: PMC2141641
- DOI: 10.1083/jcb.138.3.707
CD36 mediates the In vitro inhibitory effects of thrombospondin-1 on endothelial cells
Abstract
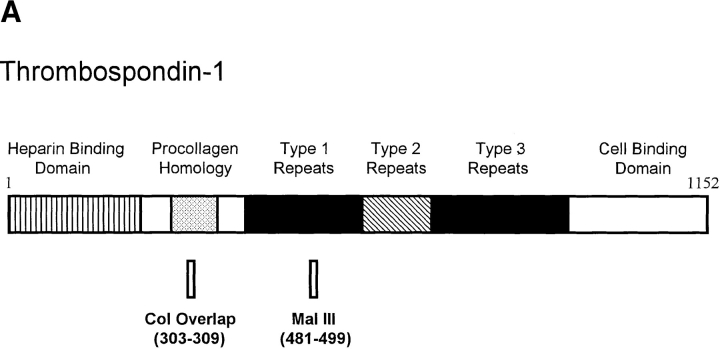
Thrombospondin-1 (TSP-1) is a naturally occurring inhibitor of angiogenesis that is able to make normal endothelial cells unresponsive to a wide variety of inducers. Here we use both native TSP-1 and small antiangiogenic peptides derived from it to show that this inhibition is mediated by CD36, a transmembrane glycoprotein found on microvascular endothelial cells. Both IgG antibodies against CD36 and glutathione-S-transferase-CD36 fusion proteins that contain the TSP-1 binding site blocked the ability of intact TSP-1 and its active peptides to inhibit the migration of cultured microvascular endothelial cells. In addition, antiangiogenic TSP-1 peptides inhibited the binding of native TSP-1 to solid phase CD36 and its fusion proteins, as well as to CD36-expressing cells. Additional molecules known to bind CD36, including the IgM anti-CD36 antibody SM, oxidized (but not unoxidized) low density lipoprotein, and human collagen 1, mimicked TSP-1 by inhibiting the migration of human microvascular endothelial cells. Transfection of CD36-deficient human umbilical vein endothelial cells with a CD36 expression plasmid caused them to become sensitive to TSP-1 inhibition of their migration and tube formation. This work demonstrates that endothelial CD36, previously thought to be involved only in adhesion and scavenging activities, may be essential for the inhibition of angiogenesis by thrombospondin-1.
Figures













References
-
- Asch AS, Silbiger S, Heimer E, Nachman RL. Thrombospondin sequence motif (CSVTCG) is responsible for CD36 binding. Biochem Biophys Res Commun. 1992;182:1208–1217. - PubMed
-
- Asch AS, Liu I, Briccetti FM, Barnwell JW, Kwakye-Berko F, Dokun A, Goldberger J, Pernambuco M. Analysis of CD36 binding domains: ligand specificity controlled by dephosphorylation of an ectodomain. Science (Wash DC) 1993;262:1436–1440. - PubMed
-
- Bagavandoss P, Wilkes JW. Specific inhibition of endothelial cell proliferation by thrombospondin. Biochem Biophys Res Commun. 1990;170:867–872. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
