

Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos
- PMID: 9275227
- PMCID: PMC23295
- DOI: 10.1073/pnas.94.18.9920
Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos
Abstract
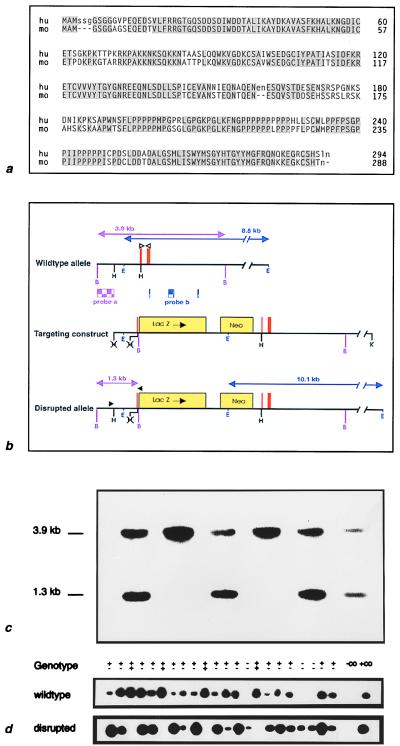
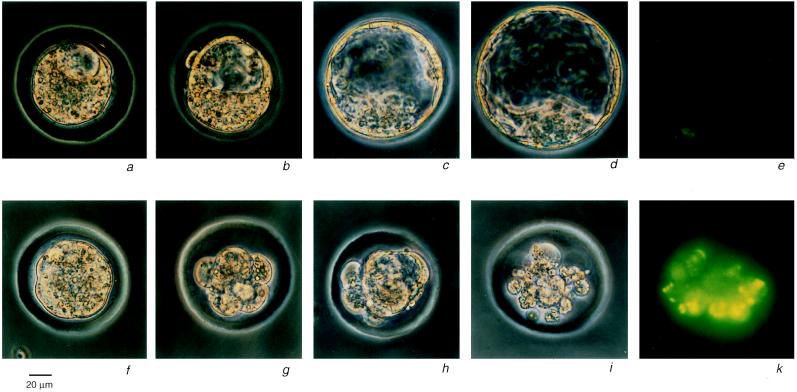
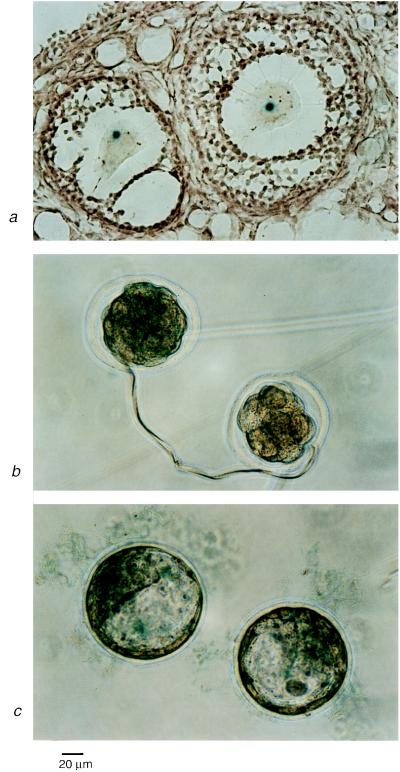
Proximal spinal muscular atrophy is an autosomal recessive human disease of spinal motor neurons leading to muscular weakness with onset predominantly in infancy and childhood. With an estimated heterozygote frequency of 1/40 it is the most common monogenic disorder lethal to infants; milder forms represent the second most common pediatric neuromuscular disorder. Two candidate genes-survival motor neuron (SMN) and neuronal apoptosis inhibitory protein have been identified on chromosome 5q13 by positional cloning. However, the functional impact of these genes and the mechanism leading to a degeneration of motor neurons remain to be defined. To analyze the role of the SMN gene product in vivo we generated SMN-deficient mice. In contrast to the human genome, which contains two copies, the mouse genome contains only one SMN gene. Mice with homozygous SMN disruption display massive cell death during early embryonic development, indicating that the SMN gene product is necessary for cellular survival and function.
Figures
References
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
