

DNA methylation inhibits elongation but not initiation of transcription in Neurospora crassa
- PMID: 9308966
- PMCID: PMC316521
- DOI: 10.1101/gad.11.18.2383
DNA methylation inhibits elongation but not initiation of transcription in Neurospora crassa
Abstract
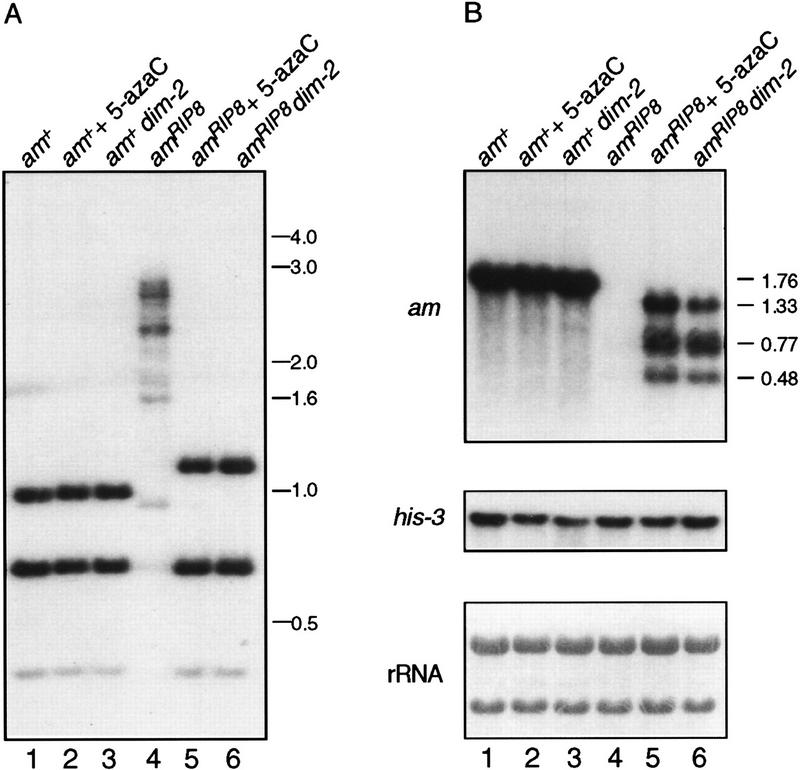
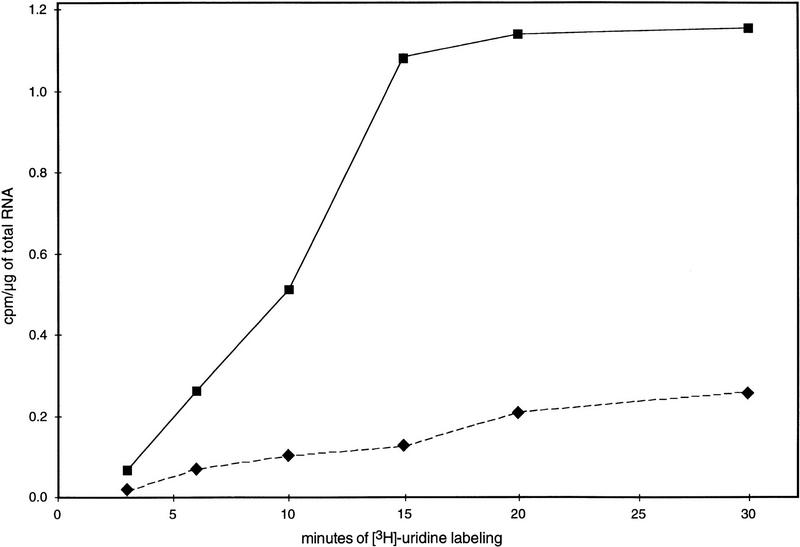
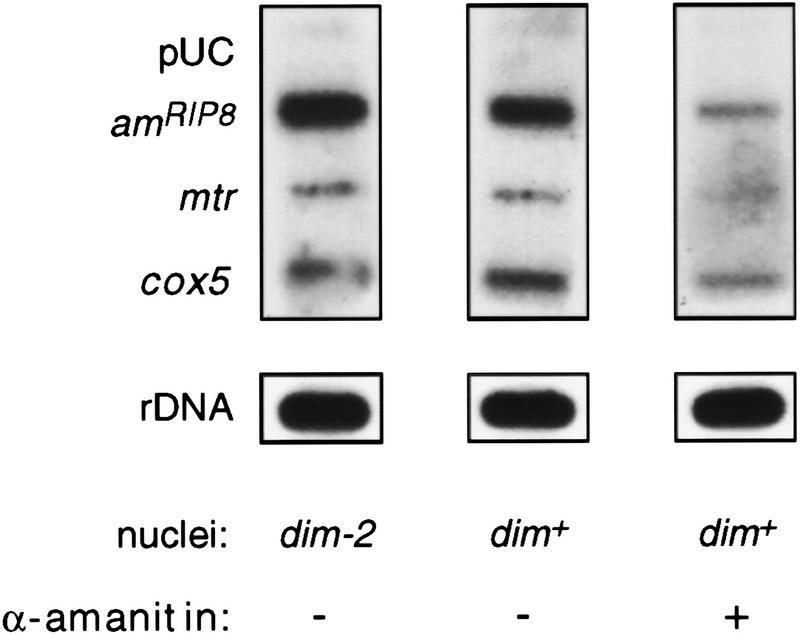
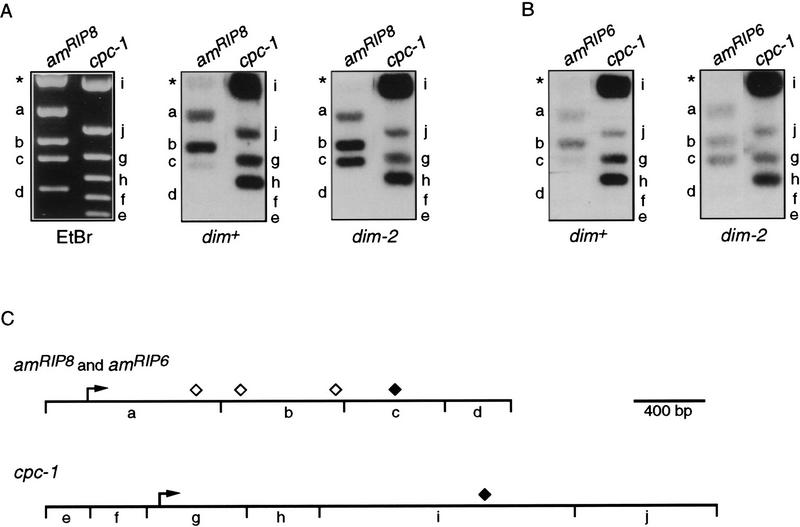
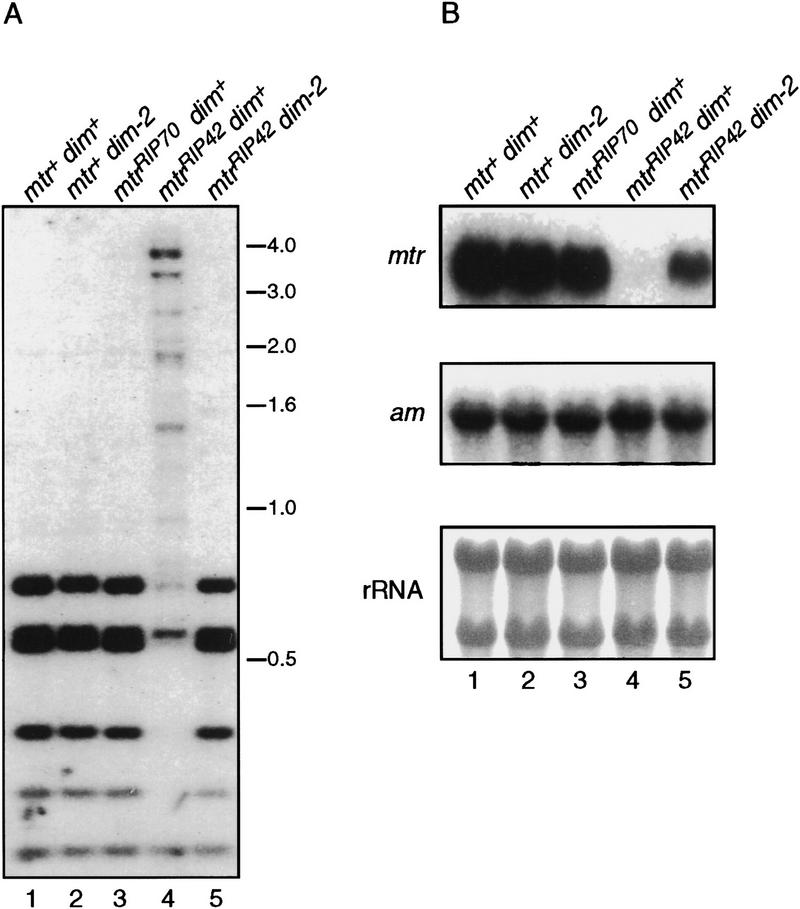
In plants, animals, and fungi, DNA methylation is frequently associated with gene silencing, yet little is known about the role of the methylation in silencing. In Neurospora crassa, repeated sequences are silenced by repeat-induced point mutation (RIP) and genes that have suffered numerous GC --> AT mutations by RIP are typically methylated at remaining cytosines. We investigated possible effects on transcription from methylation associated with RIP by taking advantage of 5-azacytidine, which prevents most methylation in Neurospora and a dim-2 mutation that abolishes all detectable methylation. Northern analyses revealed that methylation prevents the accumulation of transcripts from genes mutated by RIP. Measurements of transcription rates in vivo showed that methylation inhibits transcription severely but does not influence mRNA stability. Results of nuclear run-on experiments demonstrated that transcription initiation was not significantly inhibited by the dense methylation in the promoter sequences. In contrast, methylation blocked transcription elongation in vivo.
Figures


References
-
- Bartolomei MS, Webber AL, Brunkow ME, Tilghman SM. Epigenetic mechanisms underlying the imprinting of the mouse H19 gene. Genes & Dev. 1993;7:1663–1673. - PubMed
-
- Bednarik DP, Duckett C, Kim SU, Perez VL, Griffis K, Guenther PC, Folks TM. DNA CpG methylation inhibits binding of NF-kB proteins to the HIV-1 long terminal repeat cognate DNA motifs. New Biol. 1991;3:969–976. - PubMed
-
- Beelman CA, Parker R. Degradation of mRNA in eukaryotes. Cell. 1995;81:179–183. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous