

GTP hydrolysis controls stringent selection of the AUG start codon during translation initiation in Saccharomyces cerevisiae
- PMID: 9308967
- PMCID: PMC316512
- DOI: 10.1101/gad.11.18.2396
GTP hydrolysis controls stringent selection of the AUG start codon during translation initiation in Saccharomyces cerevisiae
Abstract
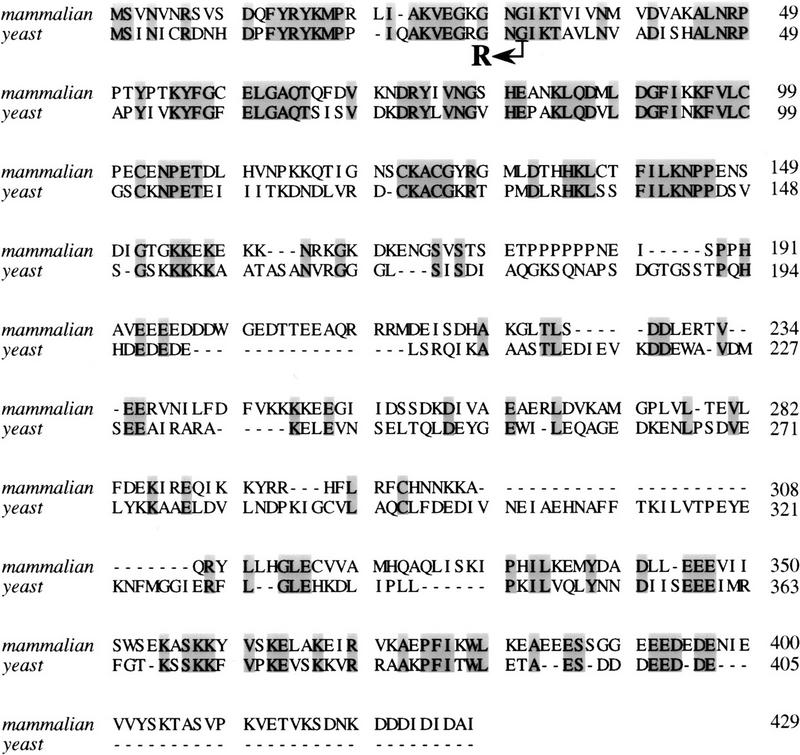
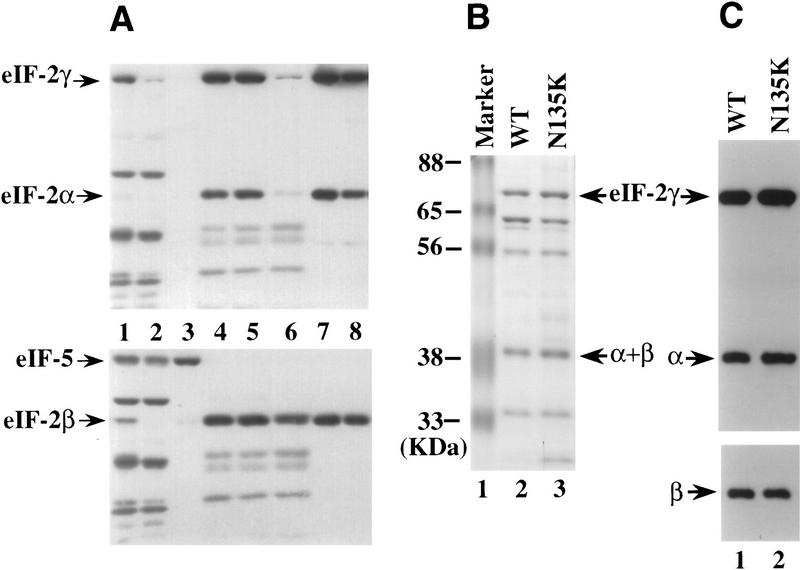
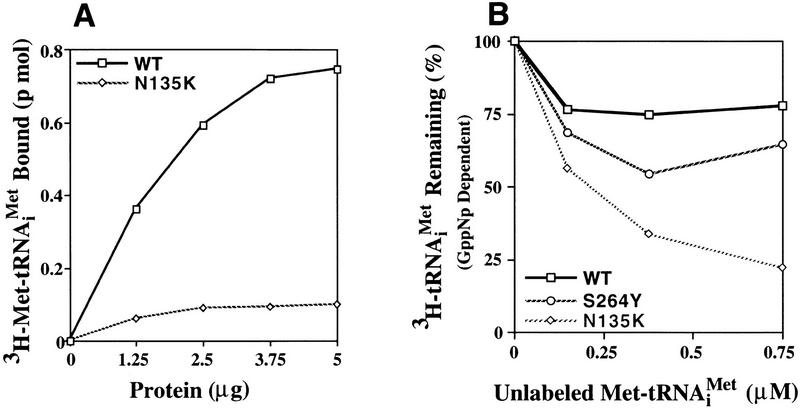
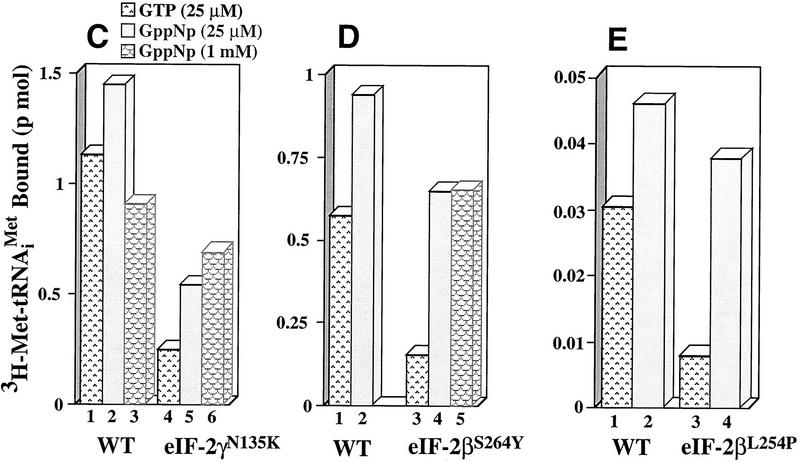
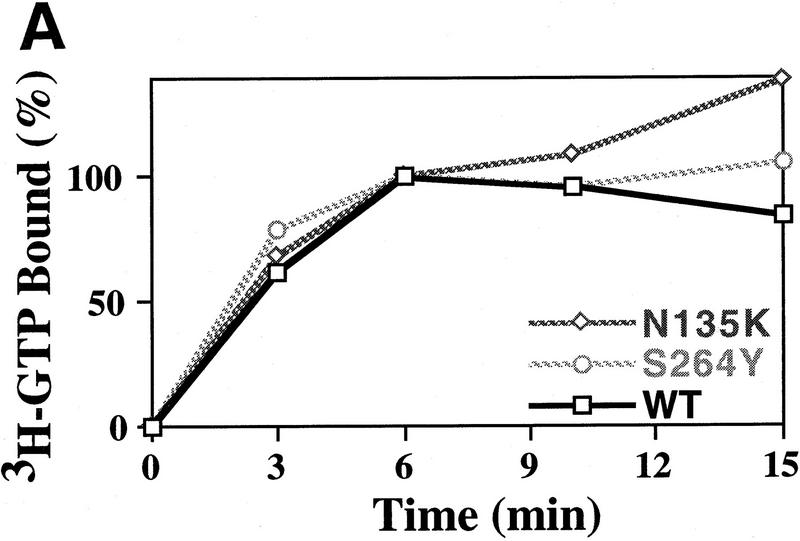
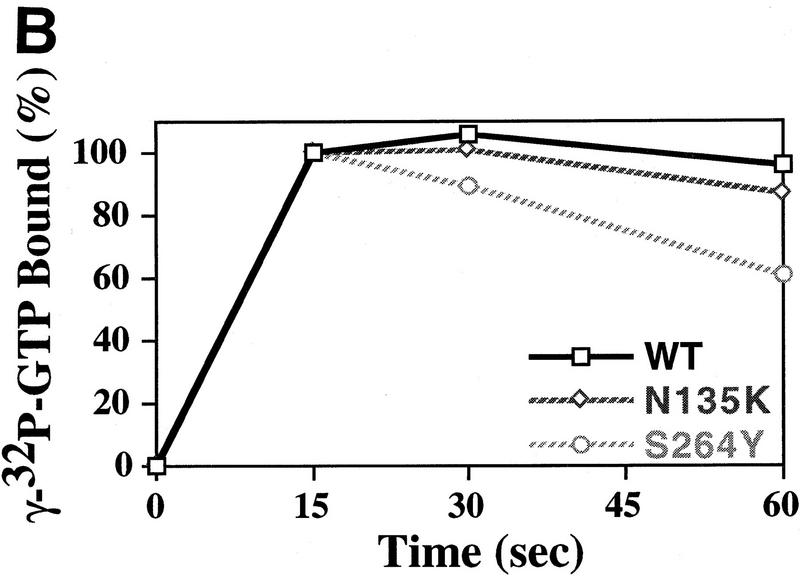
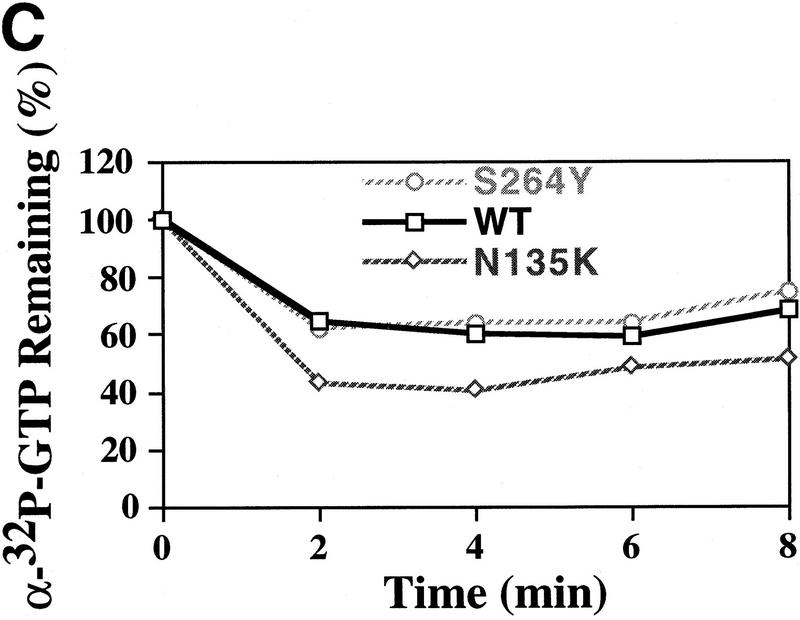
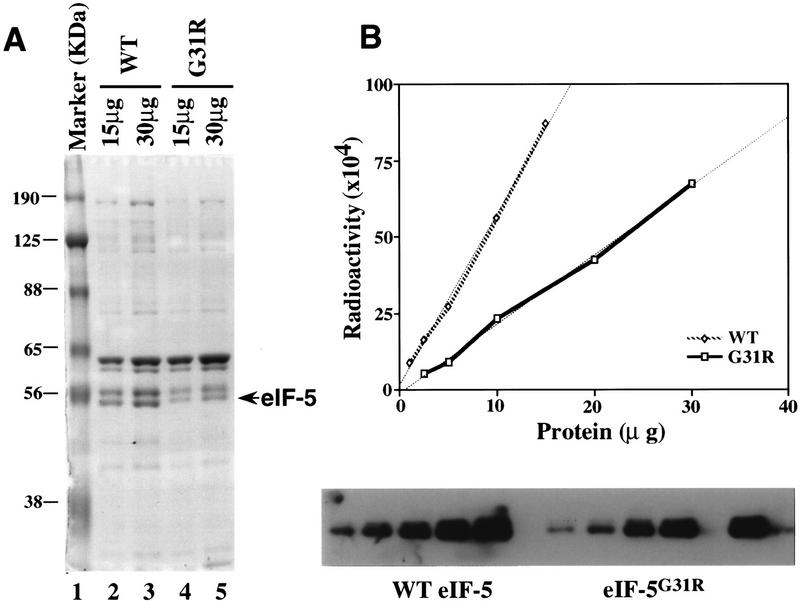
We have isolated and characterized two suppressor genes, SUI4 and SUI5, that can initiate translation in the absence of an AUG start codon at the HIS4 locus in Saccharomyces cerevisiae. Both suppressor genes are dominant in diploid cells and lethal in haploid cells. The SUI4 suppressor gene is identical to the GCD11 gene, which encodes the gamma subunit of the eIF-2 complex and contains a mutation in the G2 motif, one of the four signature motifs that characterizes this subunit to be a G-protein. The SUI5 suppressor gene is identical to the TIF5 gene that encodes eIF-5, a translation initiation factor known to stimulate the hydrolysis of GTP bound to eIF-2 as part of the 43S preinitiation complex. Purified mutant eIF-5 is more active in stimulating GTP hydrolysis in vitro than wild-type eIF-5, suggesting that an alteration of the hydrolysis rate of GTP bound to the 43S preinitiation complex during ribosomal scanning allows translation initiation at a non-AUG codon. Purified mutant eIF-2gamma complex is defective in ternary complex formation and this defect correlates with a higher rate of dissociation from charged initiator-tRNA in the absence of GTP hydrolysis. Biochemical characterization of SUI3 suppressor alleles that encode mutant forms of the beta subunit of eIF-2 revealed that these mutant eIF-2 complexes have a higher intrinsic rate of GTP hydrolysis, which is eIF-5 independent. All of these biochemical defects result in initiation at a UUG codon at the his4 gene in yeast. These studies in light of other analyses indicate that GTP hydrolysis that leads to dissociation of eIF-2 x GDP from the initiator-tRNA in the 43S preinitiation complex serves as a checkpoint for a 3-bp codon/anticodon interaction between the AUG start codon and the initiator-tRNA during the ribosomal scanning process.
Figures
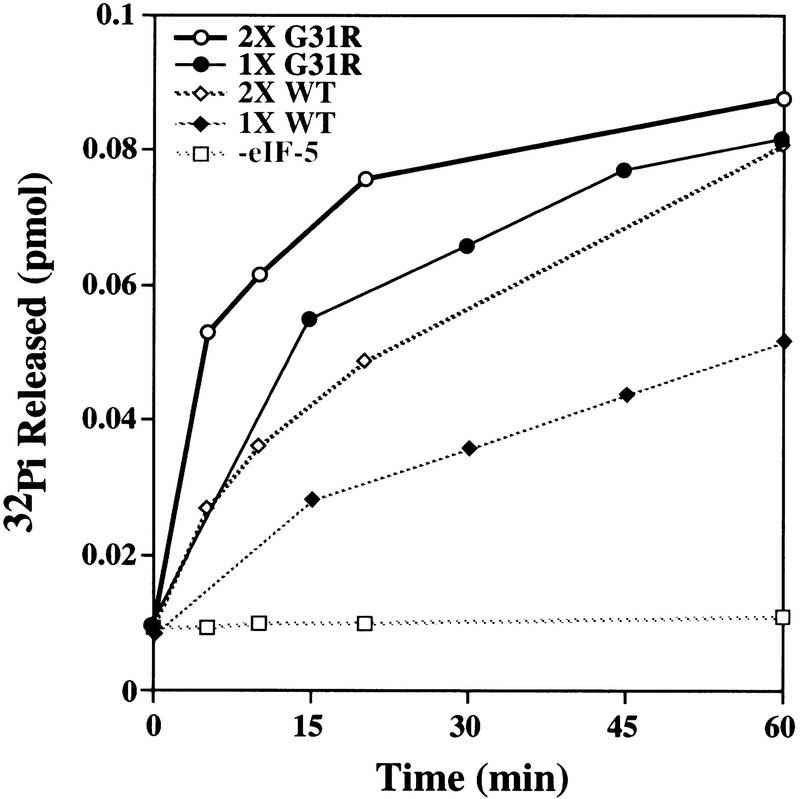
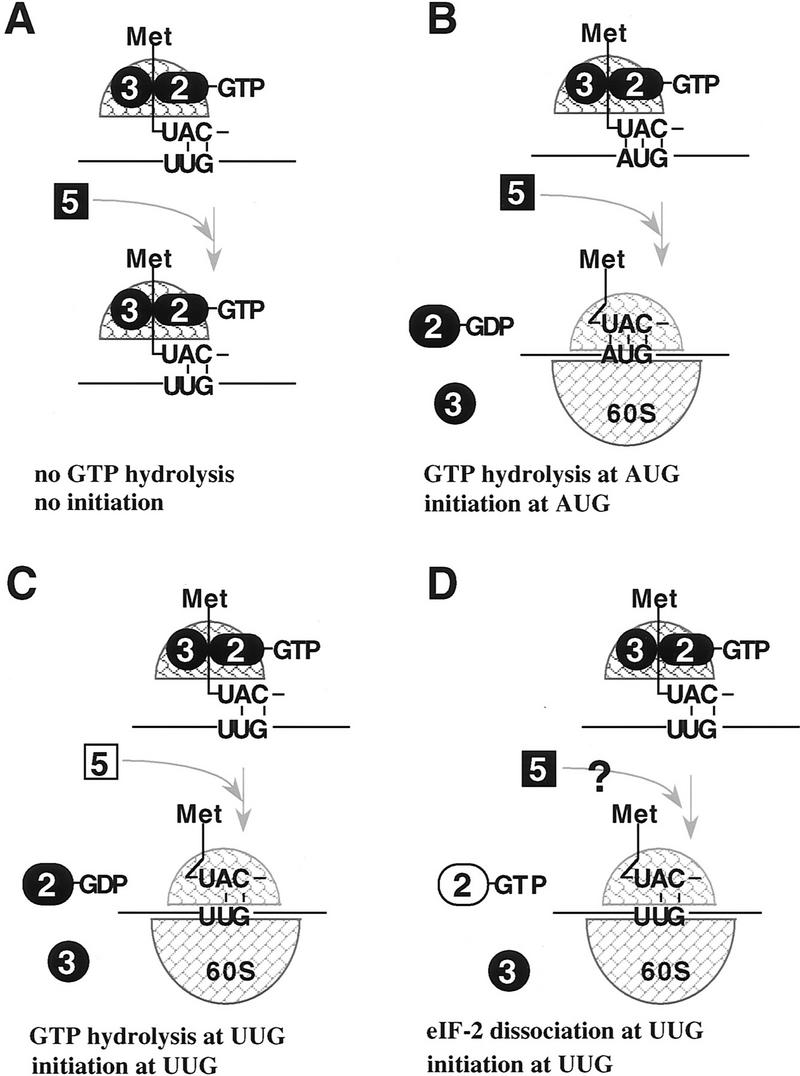
 ) Initiator tRNA; (➋) eIF-2; (➌) eIF-3; (
) Initiator tRNA; (➋) eIF-2; (➌) eIF-3; ( ) eIF-5; (
) eIF-5; ( ) the 40S ribosomal subunit; (
) the 40S ribosomal subunit; ( ) the 60S ribosomal subunit.
) the 60S ribosomal subunit.References
-
- Balch WE. Small GTP-binding proteins in vesicular transport. Trends Biochem Sci. 1990;15:473–477. - PubMed
-
- Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: Conserved structure and molecular mechanism. Nature. 1991;349:117–127. - PubMed
-
- Bushman JL, Foiani M, Cigan AM, Paddon CJ, Hinnebusch AG. Guanine nucleotide exchange factor for eukaryotic translation initiation factor 2 in Saccharomyces cerevisiae: Interactions between the essential subunits GCD2, GCD6 and GCD7 and the regulatory subunit GCN3. Mol Cell Biol. 1993b;13:4618–4631. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous