

Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/Ern4p that activates the unfolded protein response
- PMID: 9348528
- PMCID: PMC25627
- DOI: 10.1091/mbc.8.10.1845
Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/Ern4p that activates the unfolded protein response
Abstract
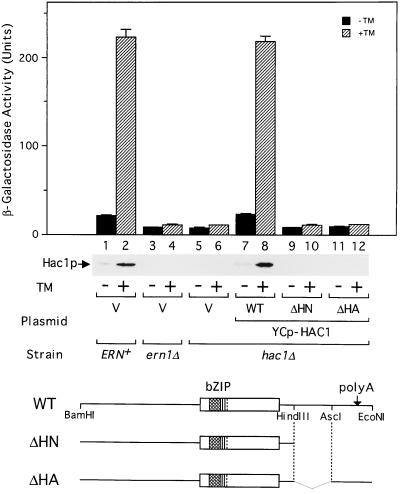
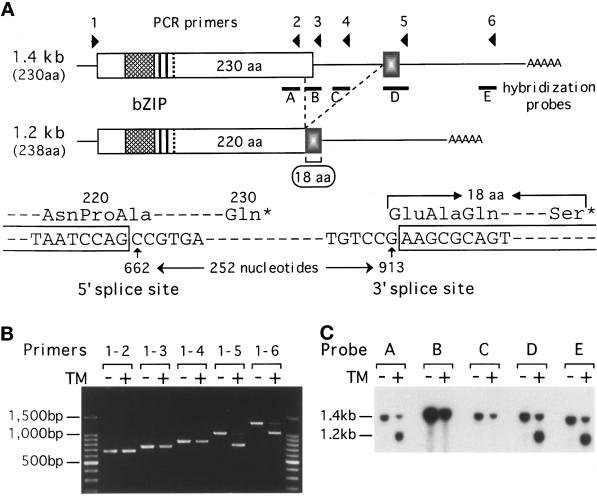
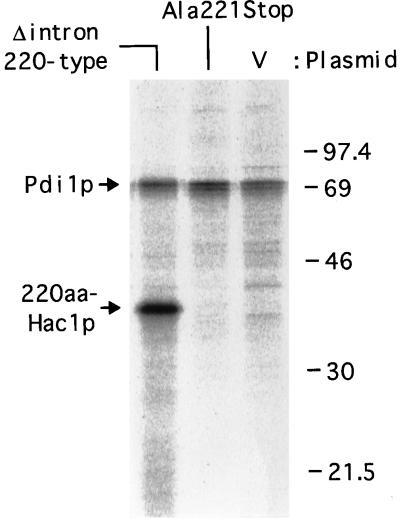
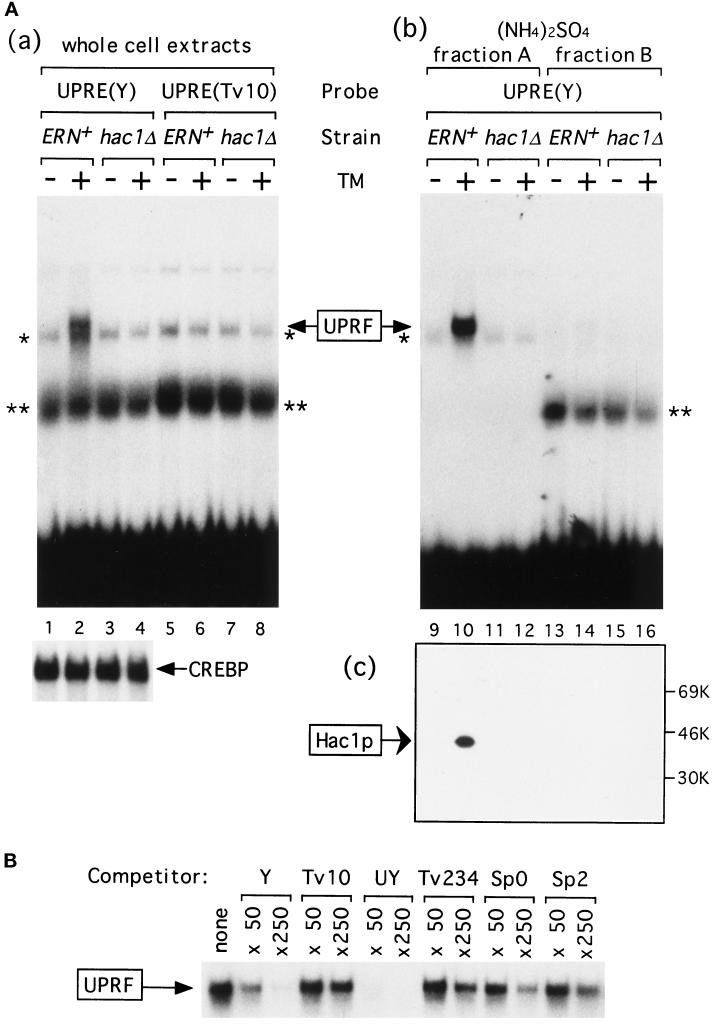
An intracellular signaling from the endoplasmic reticulum (ER) to the nucleus, called the unfolded protein response (UPR), is activated when unfolded proteins are accumulated in the ER under a variety of stress conditions ("ER stress"). We and others recently identified Hac1p/Ern4p as a transcription factor responsible for the UPR in Saccharomyces cerevisiae. It was further reported that Hac1p (238 aa) is detected only in ER-stressed cells, and its expression is mediated by unconventional splicing of HAC1 precursor mRNA. The splicing replaces the C-terminal portion of Hac1p; it was proposed that precursor mRNA is also translated but the putative product of 230 aa is rapidly degraded by the ubiquitin-proteasome pathway. We have identified and characterized the same regulated splicing and confirmed its essential features. Contrary to the above proposal, however, we find that the 238-aa product of mature mRNA and the 230-aa-type protein tested are highly unstable with little of no difference in stability. Furthermore, we demonstrate that the absence of Hac1p in unstressed cells is due to the lack of translation of precursor mRNA. We conclude that Hac1p is synthesized as the result of ER stress-induced mRNA splicing, leading to activation of the UPR.
Figures






References
-
- Brodsky JL, McCracken AA. ER-associated and proteasome-mediated protein degradation: how two topologically restricted events came together. Trends Cell Biol. 1997;7:151–156. - PubMed
-
- Chen P, Johnson P, Sommer T, Jentsch S, Hochstrasser M. Multiple ubiquitin-conjugating enzymes participate in the in vivo degradation of the yeast MATα2 repressor. Cell. 1993;74:357–69. - PubMed
-
- Cox JS, Shamu CE, Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. 1993;73:1197–1206. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
