

Regulation of the replication initiator protein p65cdc18 by CDK phosphorylation
- PMID: 9353247
- PMCID: PMC316667
- DOI: 10.1101/gad.11.21.2767
Regulation of the replication initiator protein p65cdc18 by CDK phosphorylation
Abstract
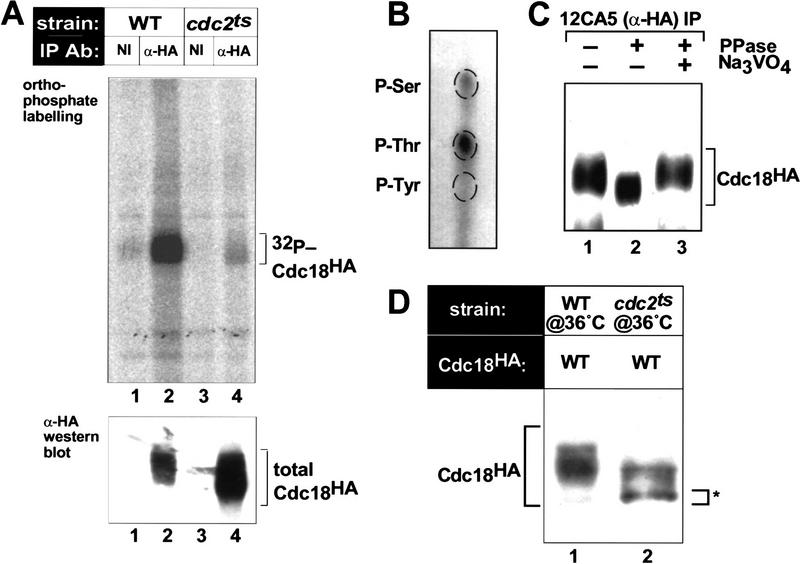
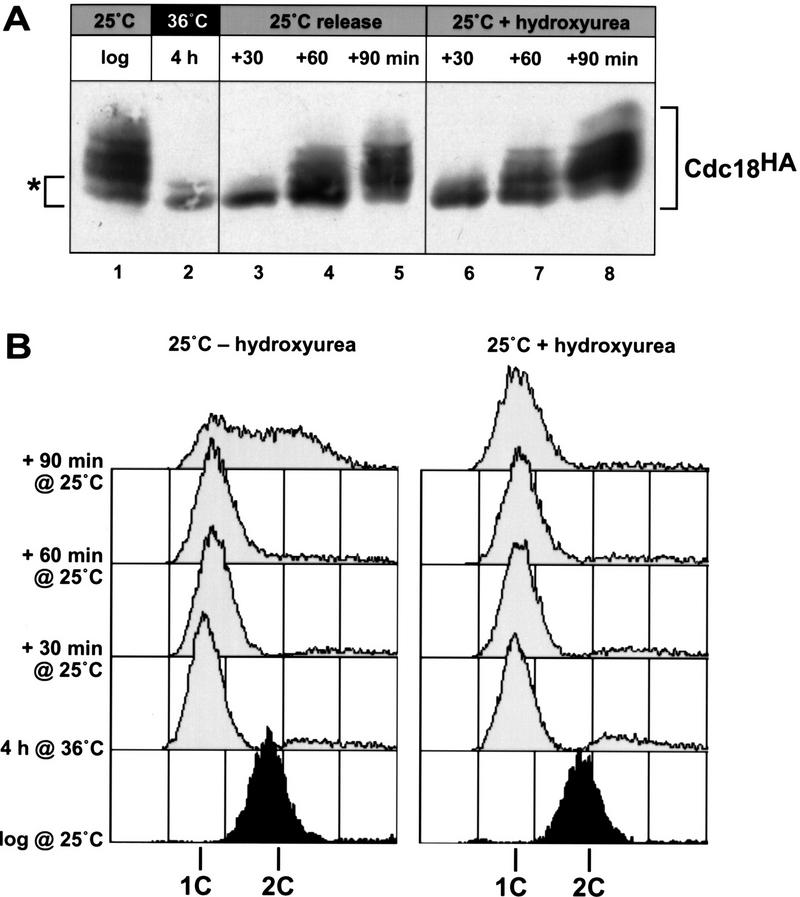
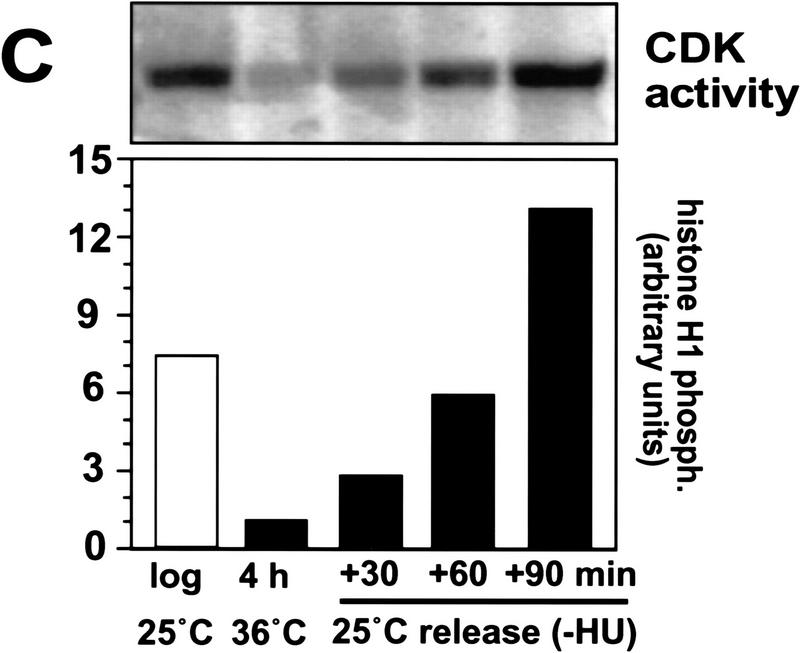
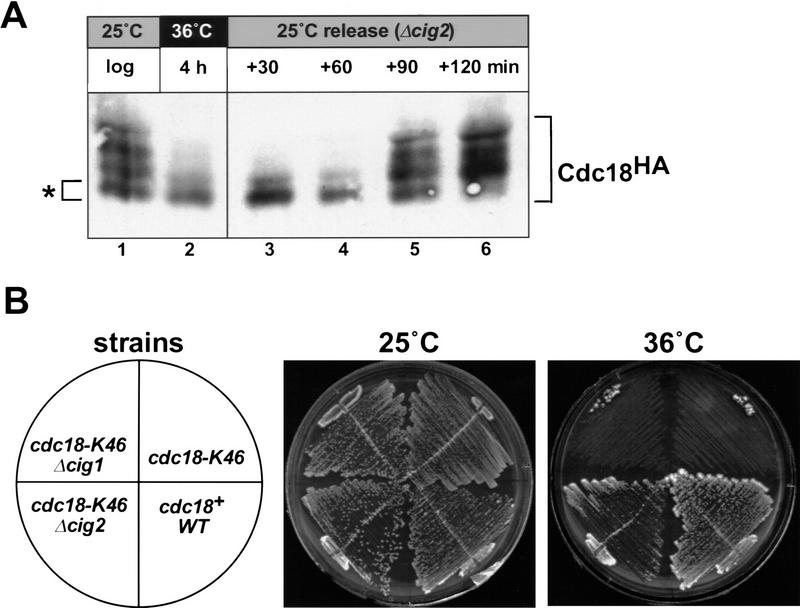
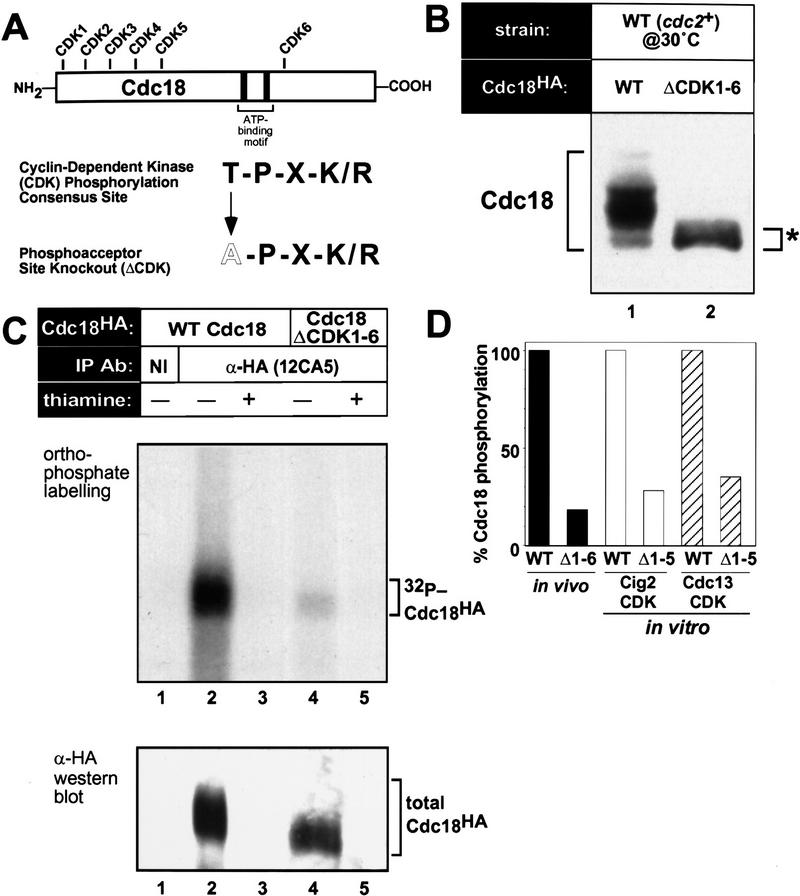
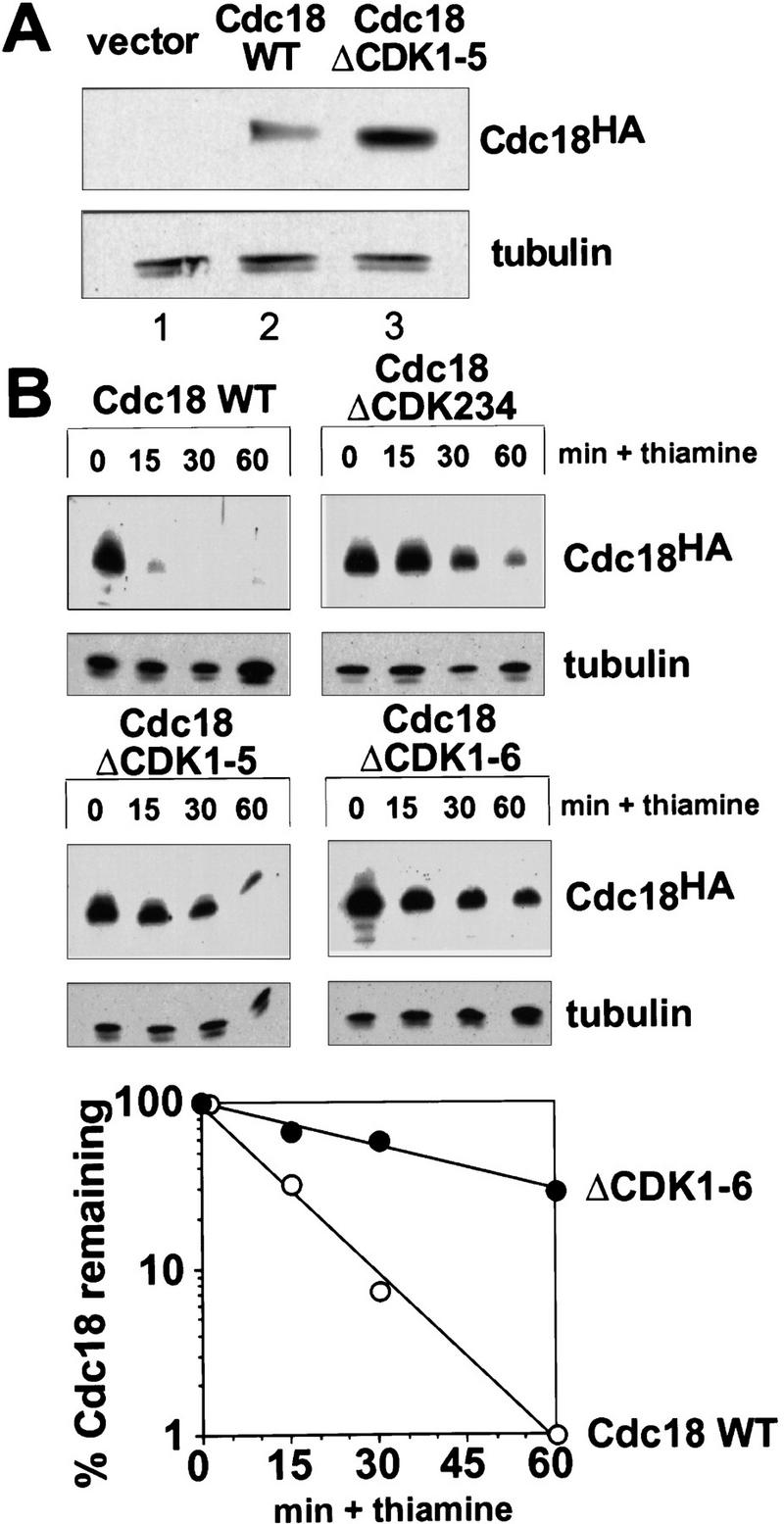
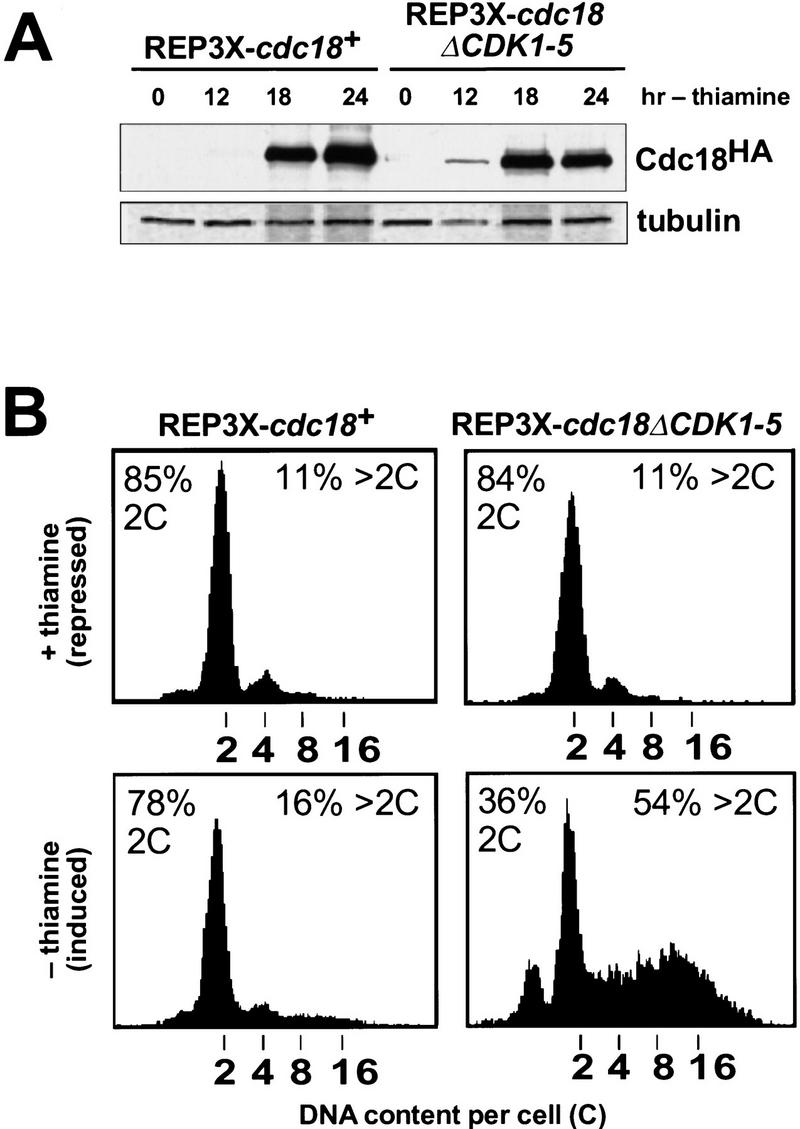
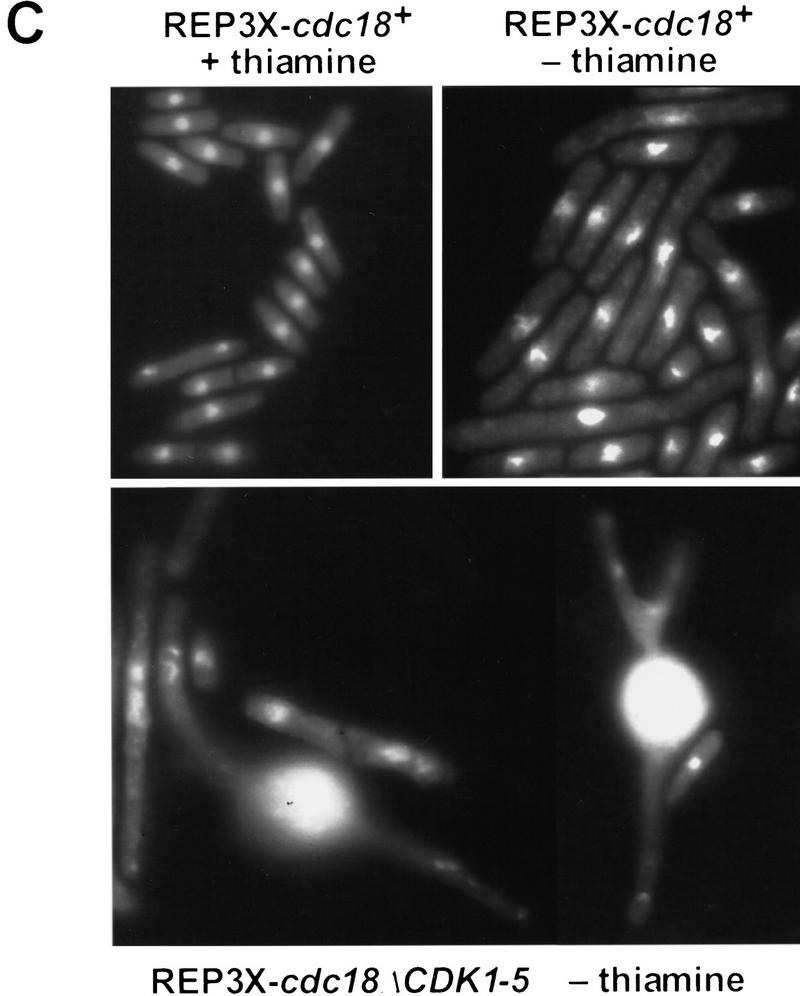
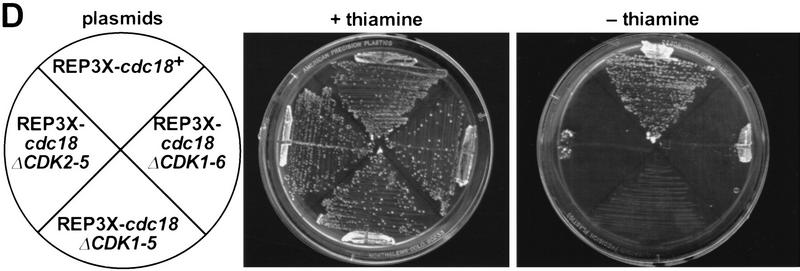
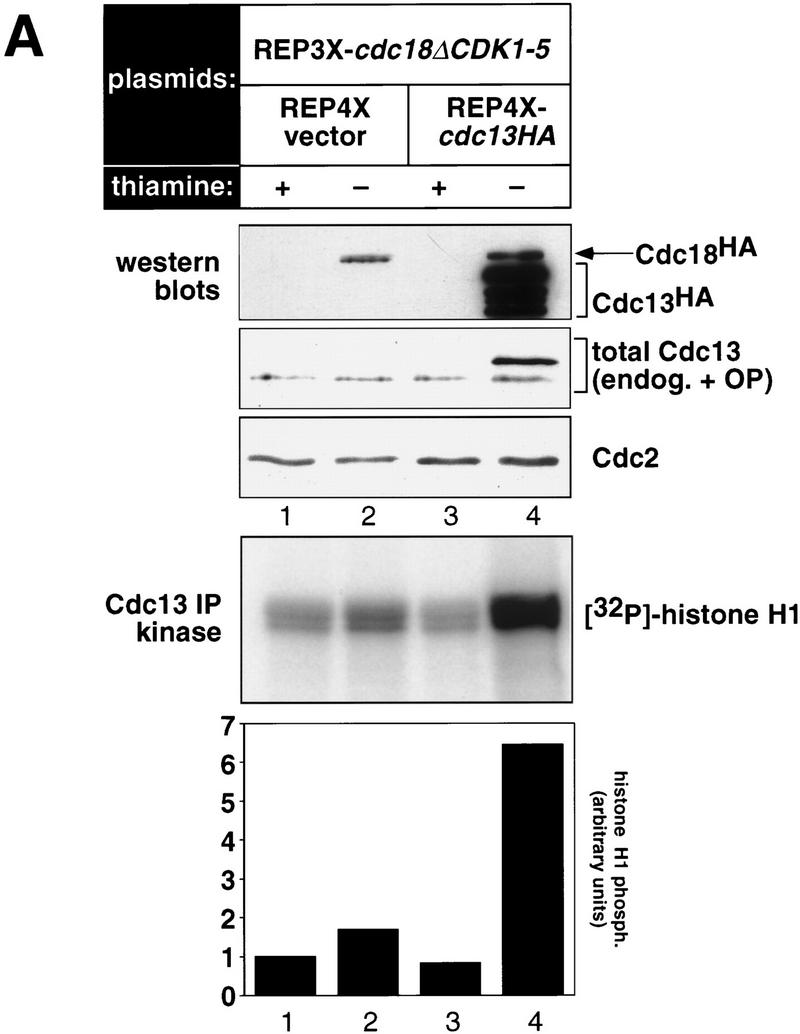
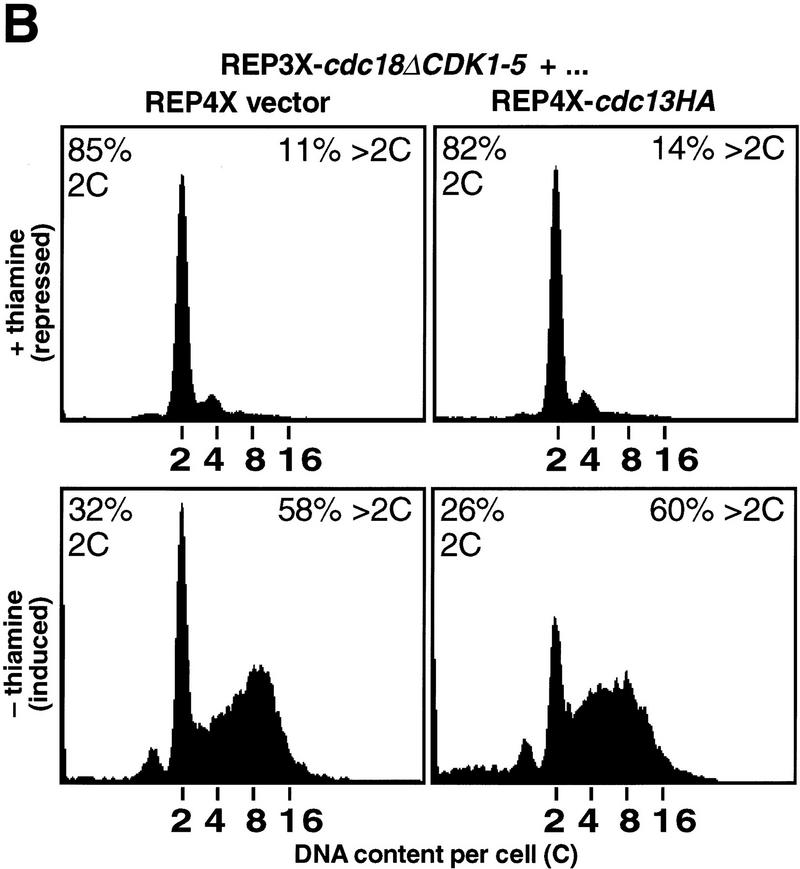
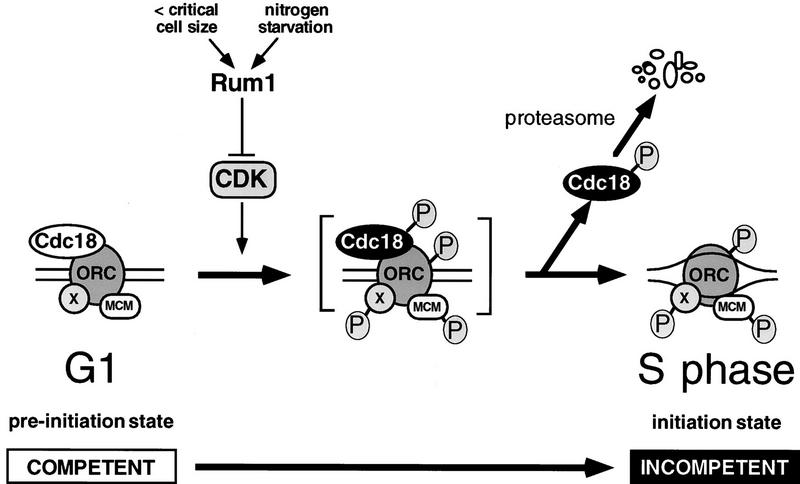
Cyclin-dependent kinases (CDKs) promote the initiation of DNA replication and prevent reinitiation before mitosis, presumably through phosphorylation of key substrates at origins of replication. In fission yeast, the p65cdc18 protein is required to initiate DNA replication and interacts with the origin recognition complex (ORC) and the p34cdc2 CDK. Here we report that p65cdc18 becomes highly phosphorylated as cells undergo the G1 --> S phase transition. This modification is dependent on p34cdc2 protein kinase activity, as well as six consensus CDK phosphorylation sites within the p65cdc18 polypeptide. Genetic interactions between cdc18+ and the S-phase cyclin cig2+ suggest that CDK-dependent phosphorylation antagonizes cdc18+ function in vivo. Using site-directed mutagenesis, we show that phosphorylation at CDK consensus sites directly targets p65cdc18 for rapid degradation and inhibits its replication activity, as strong expression of a constitutively hypophosphorylated mutant form of p65cdc18 results in large amounts of DNA over-replication in vivo. Furthermore, the over-replication phenotype produced by this mutant p65cdc18 is resistant to increased mitotic cyclin/CDK activity, a known inhibitor of over-replication. Therefore, p65cdc18 is the first example of a cellular initiation factor directly regulated in vivo by CDK-dependent phosphorylation and proteolysis. Regulation of p65cdc18 by CDK phosphorylation is likely to contribute to the CDK-driven "replication switch" that restricts initiation at eukaryotic origins to once per cell cycle.
Figures

References
-
- Ayscough K, Warren G. Inhibition of protein synthesis disrupts the Golgi apparatus in the fission yeast, Schizosaccharomyces pombe. Yeast. 1994;10:1–11. - PubMed
-
- Basi G, Schmid E, Maundrell K. TATA box mutations in the Schizosaccharomyces pombe nmt1 promoter affect transcription efficiency but not the transcription start point or thiamine repressibility. Gene. 1993;123:131–136. - PubMed
-
- Bell SP, Mitchell J, Leber J, Kobayashi R, Stillman B. The multidomain structure of Orc1p reveals similarity to regulators of DNA replication and transcriptional silencing. Cell. 1995;83:563–568. - PubMed
-
- Boyle WJ, van der Geer P, Hunter T. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol. 1991;201:110–148. - PubMed
-
- Broek D, Bartlett R, Crawford K, Nurse P. Involvement of p34cdc2 in establishing the dependency of S phase on mitosis. Nature. 1991;349:388–393. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous