

A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism
- PMID: 9356497
- PMCID: PMC25055
- DOI: 10.1073/pnas.94.23.12610
A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism
Abstract
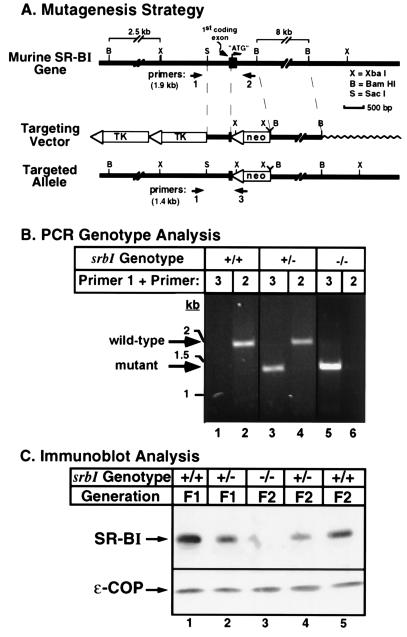
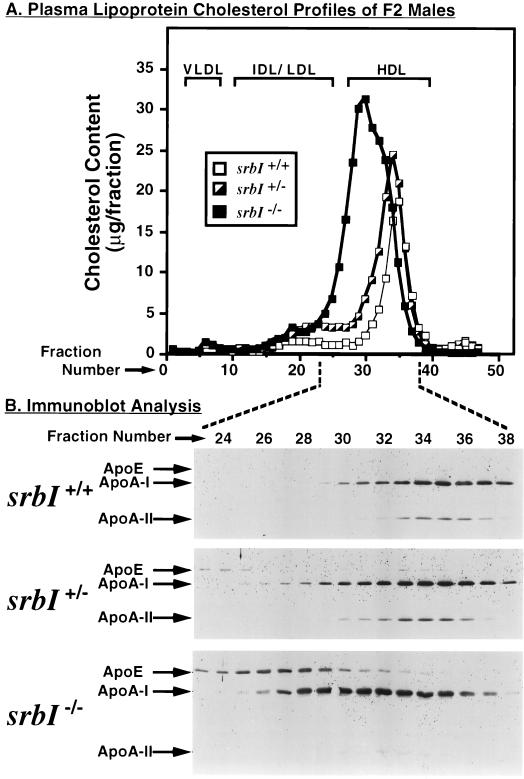
Plasma high density lipoprotein (HDL), which protects against atherosclerosis, is thought to remove cholesterol from peripheral tissues and to deliver cholesteryl esters via a selective uptake pathway to the liver (reverse cholesterol transport) and steroidogenic tissues (e.g., adrenal gland for storage and hormone synthesis). Despite its physiologic and pathophysiologic importance, the cellular metabolism of HDL has not been well defined. The class B, type I scavenger receptor (SR-BI) has been proposed to play an important role in HDL metabolism because (i) it is a cell surface HDL receptor which mediates selective cholesterol uptake in cultured cells, (ii) its physiologically regulated expression is most abundant in the liver and steroidogenic tissues, and (iii) hepatic overexpression dramatically lowers plasma HDL. To test directly the normal role of SR-BI in HDL metabolism, we generated mice with a targeted null mutation in the SR-BI gene. In heterozygous and homozygous mutants relative to wild-type controls, plasma cholesterol concentrations were increased by approximately 31% and 125%, respectively, because of the formation of large, apolipoprotein A-I (apoA-I)-containing particles, and adrenal gland cholesterol content decreased by 42% and 72%, respectively. The plasma concentration of apoA-I, the major protein in HDL, was unchanged in the mutants. This, in conjunction with the increased lipoprotein size, suggests that the increased plasma cholesterol in the mutants was due to decreased selective cholesterol uptake. These results provide strong support for the proposal that in mice the gene encoding SR-BI plays a key role in determining the levels of plasma lipoprotein cholesterol (primarily HDL) and the accumulation of cholesterol stores in the adrenal gland. If it has a similar role in controlling plasma HDL in humans, SR-BI may influence the development and progression of atherosclerosis and may be an attractive candidate for therapeutic intervention in this disease.
Figures
References
-
- Eisenberg S. J Lipid Res. 1984;25:1017–1058. - PubMed
-
- Gordon D J, Rifkin B M. N Engl J Med. 1989;321:1311–13162. - PubMed
-
- Johnson W J, Mahlberg F H, Rothblat G H, Phillips M C. Biochim Biophys Acta. 1991;1085:273–298. - PubMed
-
- Pieters M N, Schouten D, VanBerkel T J C. Biochim Biophys Acta. 1994;1225:125–134. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
