

In vitro ischemia promotes glutamate-mediated free radical generation and intracellular calcium accumulation in hippocampal pyramidal neurons
- PMID: 9364055
- PMCID: PMC6573619
- DOI: 10.1523/JNEUROSCI.17-23-09085.1997
In vitro ischemia promotes glutamate-mediated free radical generation and intracellular calcium accumulation in hippocampal pyramidal neurons
Abstract
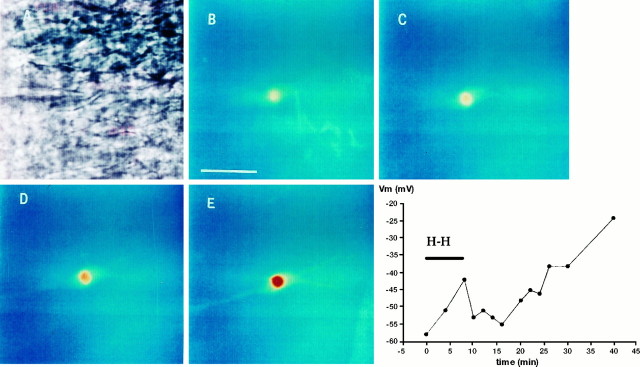
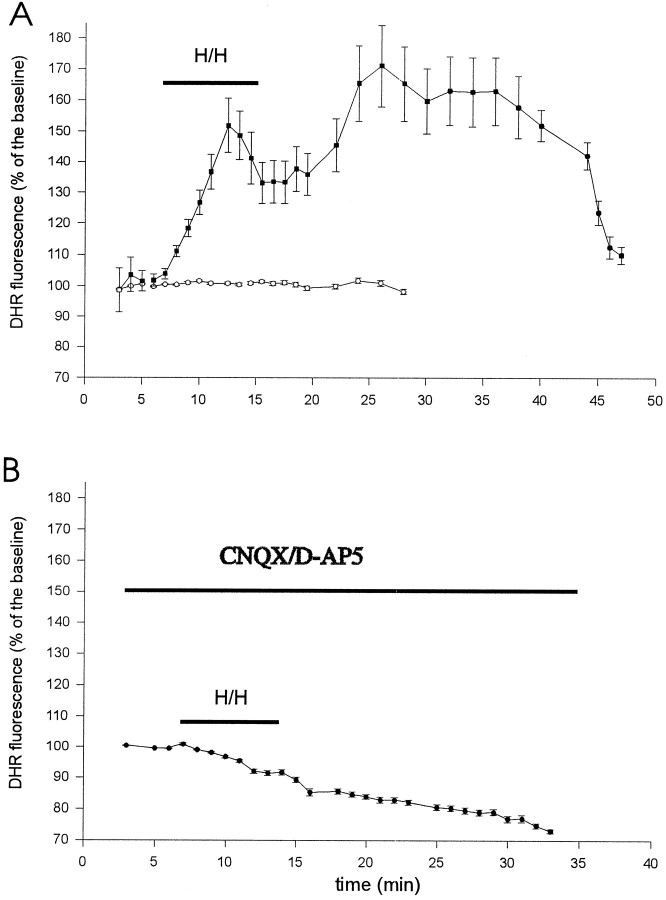
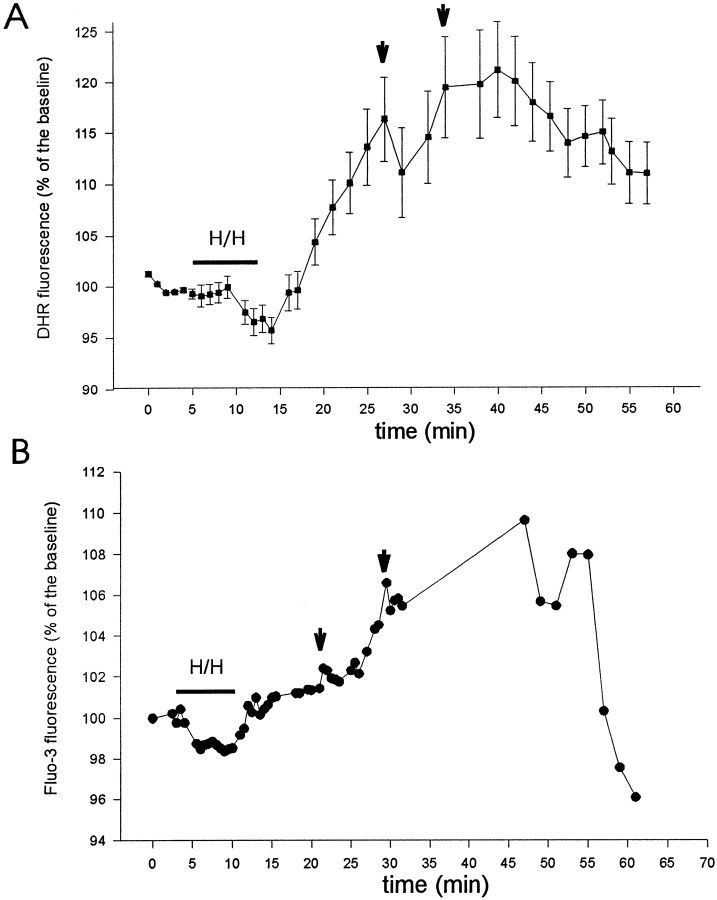
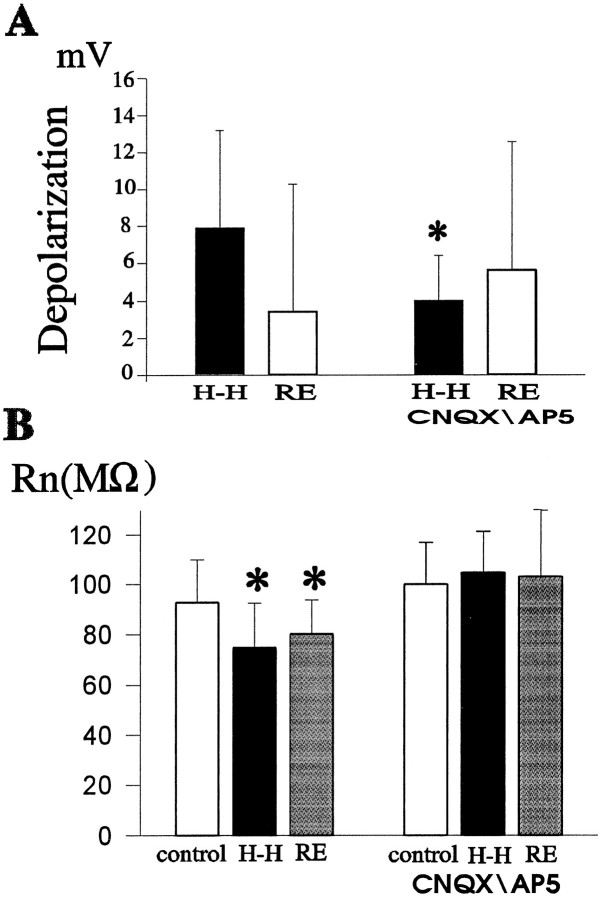
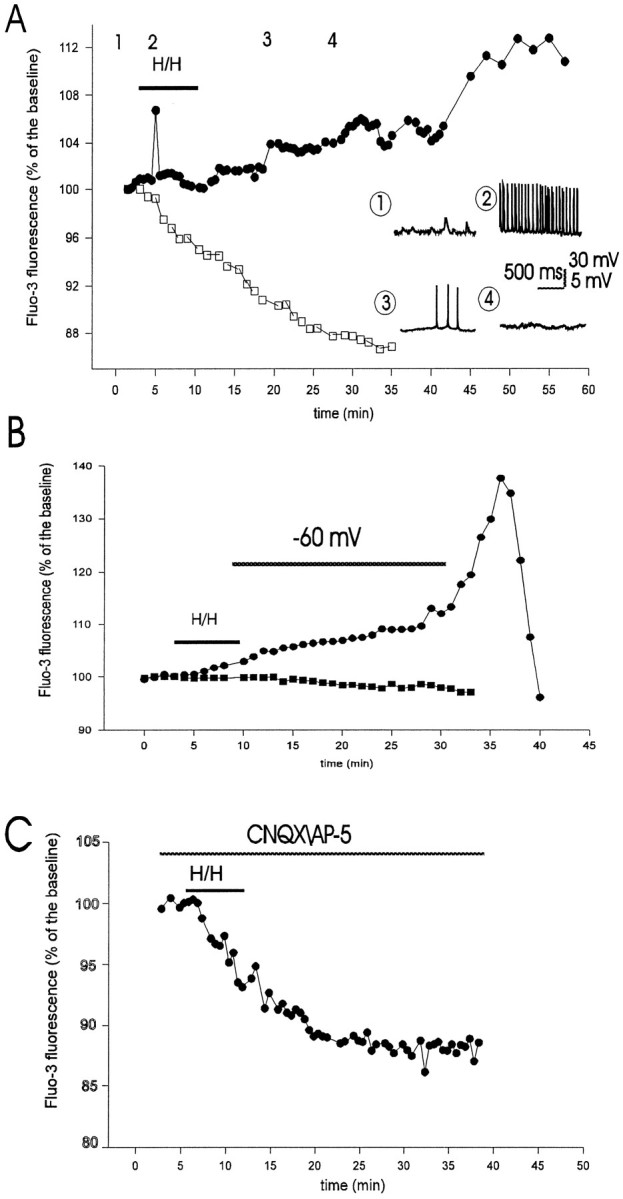
Ischemia-induced cell damage studies have revealed a complex mechanism that is thought to involve glutamate excitotoxicity, intracellular calcium increase, and free radical production. We provide direct evidence that free radical generation occurs in rat CA1 pyramidal neurons of organotypic slices subjected to a hypoxic-hypoglycemic insult. The production of free radicals is temporally correlated with intracellular calcium elevation, as measured by injection of fluo-3 in individual pyramidal cells, using patch electrodes. Free radical production (measured as changes in the fluorescence emission of dihydrorhodamine 123) peaked during reoxygenation and paralleled rising intracellular calcium. Electrophysiological whole-cell recordings revealed membrane potential depolarization and decreased input resistance during the ischemic insult. Glutamate receptor blockade resulted in decreased free radical production and markedly diminished intracellular calcium accumulation, and prevented neuronal depolarization and input resistance decrease during the ischemic episode. These results provide evidence for a direct involvement of glutamate in oxidative damage resulting from ischemic episodes.
Figures


Similar articles
-
Dynamics of intracellular calcium and free radical production during ischemia in pyramidal neurons.Free Radic Biol Med. 2001 Nov 15;31(10):1216-27. doi: 10.1016/s0891-5849(01)00705-5. Free Radic Biol Med. 2001. PMID: 11705700
-
Enhancement in activities of large conductance calcium-activated potassium channels in CA1 pyramidal neurons of rat hippocampus after transient forebrain ischemia.Brain Res. 2000 Nov 24;884(1--2):147-54. doi: 10.1016/s0006-8993(00)02923-1. Brain Res. 2000. PMID: 11082496
-
Transient forebrain ischemia induces persistent hyperactivity of large conductance Ca2+-activated potassium channels via oxidation modulation in rat hippocampal CA1 pyramidal neurons.Eur J Neurosci. 2002 Feb;15(4):779-83. doi: 10.1046/j.1460-9568.2002.01908.x. Eur J Neurosci. 2002. PMID: 11886457
-
Possible involvement of free radical formation in post-ischemic neuronal damage.Jpn J Med. 1990 Nov-Dec;29(6):677-9. doi: 10.2169/internalmedicine1962.29.677. Jpn J Med. 1990. PMID: 2101427 Review. No abstract available.
-
[Cerebral ischemia-hypoxia and biophysical mechanisms of neurodegeneration and neuroprotection effects].Fiziol Zh (1994). 2003;49(2):7-12. Fiziol Zh (1994). 2003. PMID: 12945108 Review. Ukrainian.
Cited by
-
GOLPH3 Mediated Golgi Stress Response in Modulating N2A Cell Death upon Oxygen-Glucose Deprivation and Reoxygenation Injury.Mol Neurobiol. 2016 Mar;53(2):1377-1385. doi: 10.1007/s12035-014-9083-0. Epub 2015 Jan 30. Mol Neurobiol. 2016. PMID: 25633094
-
Combination therapy with A1 receptor agonist and vitamin C improved working memory in a mouse model of global ischemia-reperfusion.Basic Clin Neurosci. 2013 Spring;4(2):111-6. Basic Clin Neurosci. 2013. PMID: 25337336 Free PMC article.
-
Glutamate receptor requirement for neuronal death from anoxia-reoxygenation: an in Vitro model for assessment of the neuroprotective effects of estrogens.Cell Mol Neurobiol. 1999 Dec;19(6):705-18. doi: 10.1023/a:1006948921855. Cell Mol Neurobiol. 1999. PMID: 10456232 Free PMC article.
-
Reperfusion accelerates acute neuronal death induced by simulated ischemia.Exp Neurol. 2007 Aug;206(2):280-7. doi: 10.1016/j.expneurol.2007.05.017. Epub 2007 Jun 2. Exp Neurol. 2007. PMID: 17599834 Free PMC article.
-
Synthesis and biological evaluation of memantine nitrates as a potential treatment for neurodegenerative diseases.Medchemcomm. 2016 Oct 20;8(1):135-147. doi: 10.1039/c6md00509h. eCollection 2017 Jan 1. Medchemcomm. 2016. PMID: 30108699 Free PMC article.
References
-
- Ankarcrona M, Dybpukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. - PubMed
-
- Barzilai A, Rahaminoff H. Stoichiometry of the sodium-calcium exchanger in nerve terminals. Biochemistry. 1987;26:6113–6118. - PubMed
-
- Cao W, Carney JM, Duchen A, Floyd RA, Chevion M. Oxygen free radical involvement in ischemia and reperfusion injury to brain. Neurosci Lett. 1988;88:233–238. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous