

Listeria monocytogenes virulence factors that stimulate endothelial cells
- PMID: 9423863
- PMCID: PMC107882
- DOI: 10.1128/IAI.66.1.232-238.1998
Listeria monocytogenes virulence factors that stimulate endothelial cells
Abstract
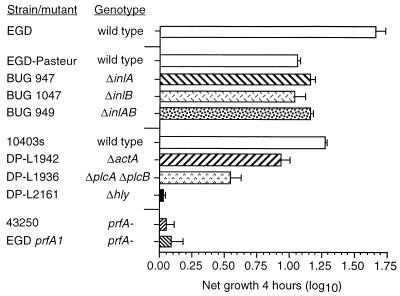
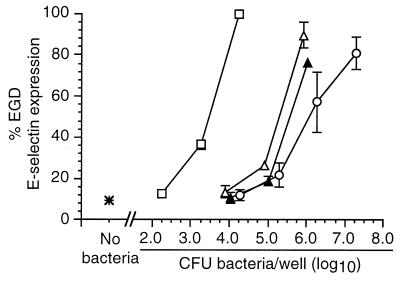
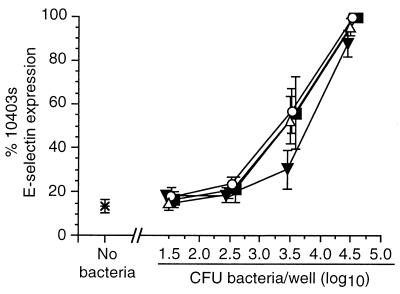
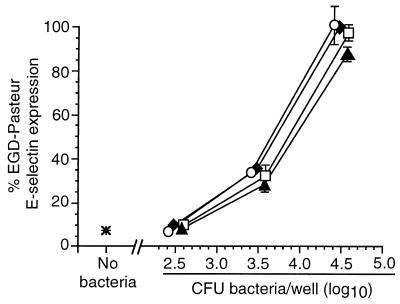
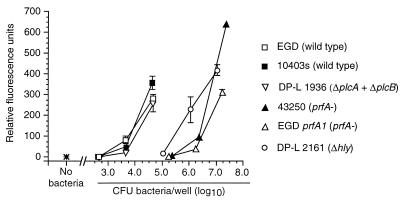
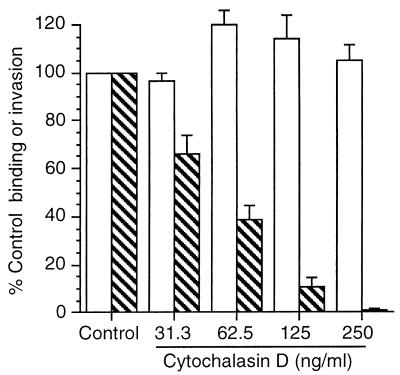
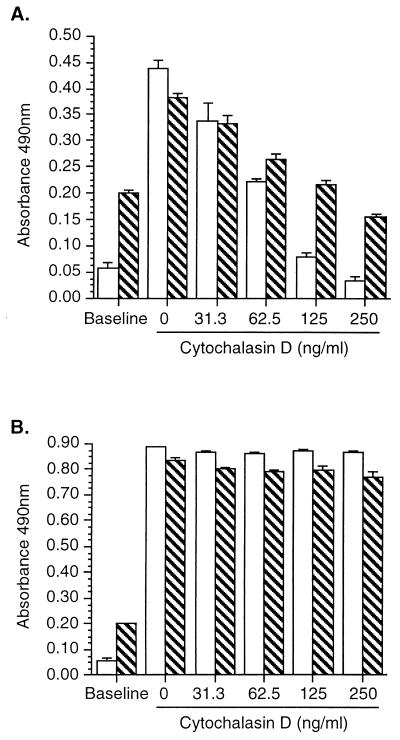
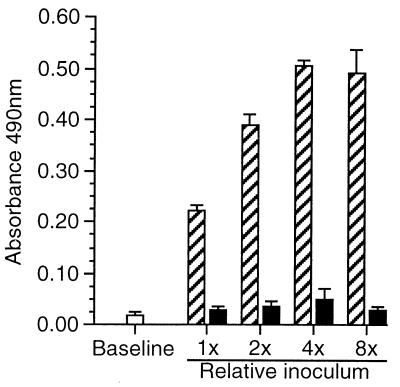
Listeria monocytogenes infection of endothelial cells upregulates surface expression of adhesion molecules and stimulates neutrophil adhesion to infected cell monolayers. The experiments presented here tested the roles of specific bacterial virulence factors as triggers for this inflammatory phenotype and function. Human umbilical vein endothelial cell (HUVEC) monolayers were infected with wild-type L. monocytogenes or L. monocytogenes mutants; then surface expression of E-selectin and neutrophil adhesion were measured. The results showed that delta hly and prfA mutants were the most crippled, requiring 100-fold more mutant bacteria than wild-type bacteria for analogous stimulation. By comparison, L. monocytogenes mutants with deletions of actA, inlA, inlB, inlAB, plcA, and plcB resembled their parent strains, and a delta plcA delta plcB mutant displayed decreased intracellular growth rate but only a minor decrease in stimulation of E-selectin or neutrophil adhesion. Other experiments showed that cytochalasin D-treated HUVEC monolayers bound bacteria, but internalization and increased surface E-selectin and intercellular adhesion molecule-1 expression were profoundly inhibited. However, cytochalasin D had no effect on the HUVEC response to stimulation with lipopolysaccharide or tumor necrosis factor alpha. These data suggest that listeriolysin O production by infecting L. monocytogenes contributes to increased expression of surface E-selectin and intercellular adhesion molecule-1, but neither it nor intracellular replication are directly responsible for this event. Nonetheless it is possible that listeriolysin O potentiates the effect(s) of an other molecule(s) that directly triggers this response. Additionally, cellular invasion by L. monocytogenes appears to be critical for initiating the HUVEC response, potentially by providing a signal which results in upregulation of the necessary bacterial genes.
Figures
References
-
- Beilke M A. Vascular endothelium in immunology and infectious disease. Rev Infect Dis. 1989;11:273–283. - PubMed
-
- Berche P. Bacteremia is required for invasion of the murine central nervous system by Listeria monocytogenes. Microb Pathog. 1995;18:323–336. - PubMed
-
- Bohne J, Sokolovic Z, Goebel W. Transcriptional regulation of prfA and PrfA-regulated virulence genes in Listeria monocytogenes. Mol Microbiol. 1994;11:1141–1150. - PubMed
-
- Bryant A E, Stevens D L. Phospholipase C and perfringolysin O from Clostridium perfringens upregulate endothelial cell-leukocyte adherence molecule 1 and intercellular leukocyte adherence molecule 1 expression and induce interleukin-8 synthesis in cultured umbilical vein endothelial cells. Infect Immun. 1996;64:358–362. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
