

Differentiation of phylogenetically related slowly growing mycobacteria based on 16S-23S rRNA gene internal transcribed spacer sequences
- PMID: 9431937
- PMCID: PMC124824
- DOI: 10.1128/JCM.36.1.139-147.1998
Differentiation of phylogenetically related slowly growing mycobacteria based on 16S-23S rRNA gene internal transcribed spacer sequences
Abstract
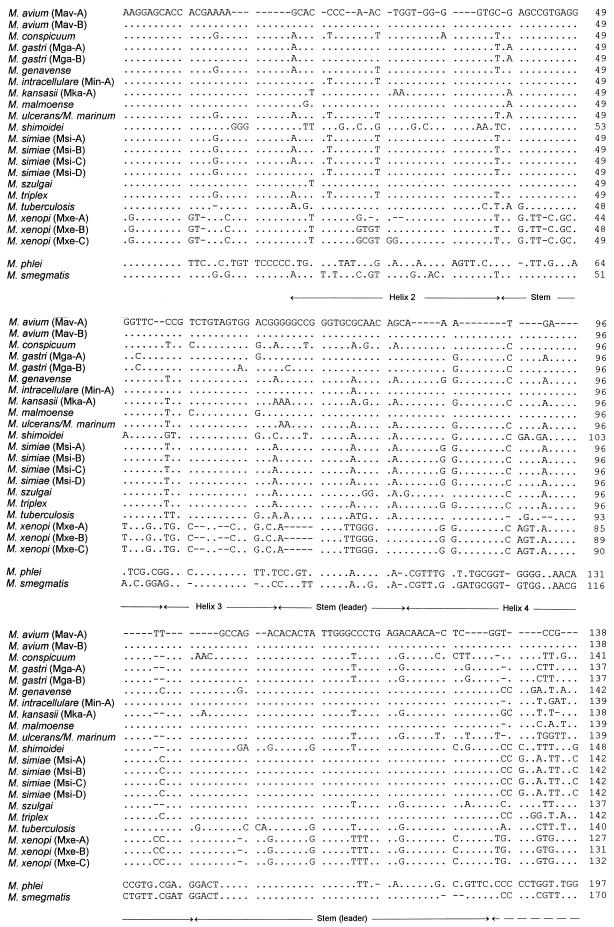
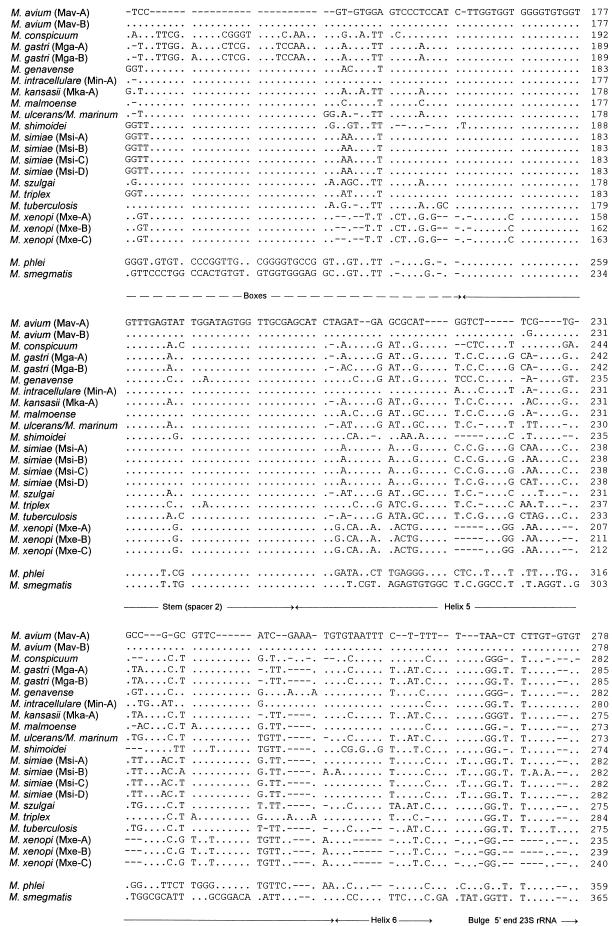
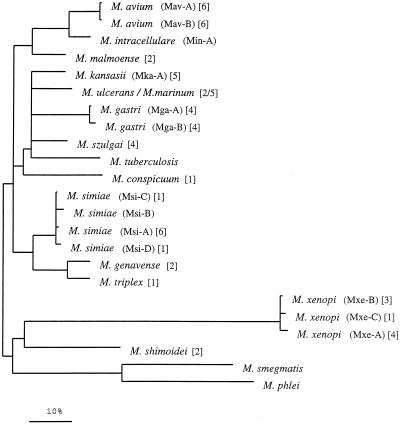
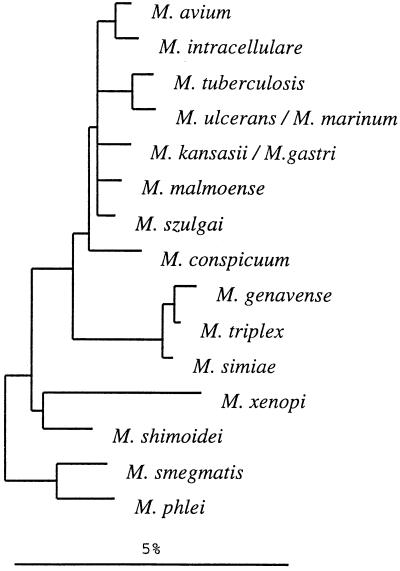
Interspecific polymorphisms of the 16S rRNA gene (rDNA) are widely used for species identification of mycobacteria. 16S rDNA sequences, however, do not vary greatly within a species, and they are either indistinguishable in some species, for example, in Mycobacterium kansasii and M. gastri, or highly similar, for example, in M. malmoense and M. szulgai. We determined 16S-23S rDNA internal transcribed spacer (ITS) sequences of 60 strains in the genus Mycobacterium representing 13 species (M. avium, M. conspicuum, M. gastri, M. genavense, M. kansasii, M. malmoense, M. marinum, M. shimoidei, M. simiae, M. szulgai, M. triplex, M. ulcerans, and M. xenopi). An alignment of these sequences together with additional sequences available in the EMBL database (for M. intracellulare, M. phlei, M. smegmatis, and M. tuberculosis) was established according to primary- and secondary-structure similarities. Comparative sequence analysis applying different treeing methods grouped the strains into species-specific clusters with low sequence divergence between strains belonging to the same species (0 to 2%). The ITS-based tree topology only partially correlated to that based on 16S rDNA, but the main branching orders were preserved, notably, the division of fast-growing from slowly growing mycobacteria, separate branching for M. simiae, M. genavense, and M. triplex, and distinct branches for M. xenopi and M. shimoidei. Comparisons of M. gastri with M. kansasii and M. malmoense with M. szulgai revealed ITS sequence similarities of 93 and 88%, respectively. M. marinum and M. ulcerans possessed identical ITS sequences. Our results show that ITS sequencing represents a supplement to 16S rRNA gene sequences for the differentiation of closely related species. Slowly growing mycobacteria show a high sequence variation in the ITS; this variation has the potential to be used for the development of probes as a rapid approach to mycobacterial identification.
Figures
References
-
- Alcaide F, Richter I, Bernasconi C, Springer B, Hagenau C, Schulze-Röbbecke R, Tortoli E, Martín R, Böttger E, Telenti A. Heterogeneity and clonality among isolates of Mycobacterium kansasii: implications for epidemiological and pathogenicity studies. J Clin Microbiol. 1997;35:1959–1964. - PMC - PubMed
-
- Barry T, Colleran G, Glennon M, Dunican L K, Gannon F. The 16S/23S ribosomal spacer region as a target for DNA probes to identify eubacteria. PCR Methods Appl. 1991;1:51–56. - PubMed
-
- De Smet A L, Brown I N, Yates M, Ivanyi J. Ribosomal internal transcribed spacers are identical among Mycobacterium avium-intracellulare complex isolates from AIDS patients, but vary among isolates from elderly pulmonary disease patients. Microbiology. 1995;141:2739–2747. - PubMed
-
- Emler S, Ninet B, Rohner P, Auckenthaler R, Jäger D, Hirschel B. Molecular basis for cross-reactivity between a strain of Mycobacterium terrae and DNA probes for Mycobacterium tuberculosis complex. Eur J Microbiol Infect Dis. 1995;14:627–629. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
