

The atomic model of the human protective protein/cathepsin A suggests a structural basis for galactosialidosis
- PMID: 9435242
- PMCID: PMC18470
- DOI: 10.1073/pnas.95.2.621
The atomic model of the human protective protein/cathepsin A suggests a structural basis for galactosialidosis
Abstract
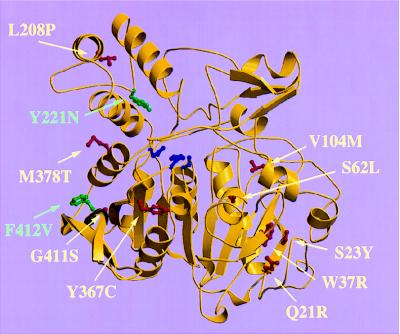
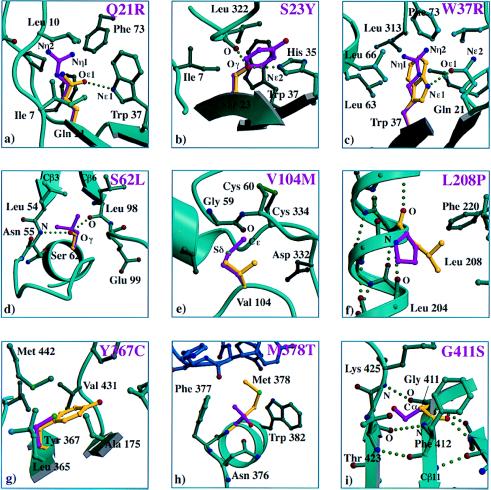
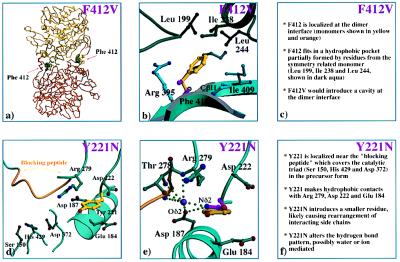
Human protective protein/cathepsin A (PPCA), a serine carboxypeptidase, forms a multienzyme complex with beta-galactosidase and neuraminidase and is required for the intralysosomal activity and stability of these two glycosidases. Genetic lesions in PPCA lead to a deficiency of beta-galactosidase and neuraminidase that is manifest as the autosomal recessive lysosomal storage disorder galactosialidosis. Eleven amino acid substitutions identified in mutant PPCAs from clinically different galactosialidosis patients have now been modeled in the three-dimensional structure of the wild-type enzyme. Of these substitutions, 9 are located in positions likely to alter drastically the folding and stability of the variant protein. In contrast, the other 2 mutations that are associated with a more moderate clinical outcome and are characterized by residual mature protein appeared to have a milder effect on protein structure. Remarkably, none of the mutations occurred in the active site or at the protein surface, which would have disrupted the catalytic activity or protective function. Instead, analysis of the 11 mutations revealed a substantive correlation between the effect of the amino acid substitution on the integrity of protein structure and the general severity of the clinical phenotype. The high incidence of PPCA folding mutants in galactosialidosis reflects the fact that a single point mutation is unlikely to affect both the beta-galactosidase and the neuraminidase binding sites of PPCA at the same time to produce the double glycosidase deficiency. Mutations in PPCA that result in defective folding, however, disrupt every function of PPCA simultaneously.
Figures
Similar articles
-
Stable expression of protective protein/cathepsin A-green fluorescent protein fusion genes in a fibroblastic cell line from a galactosialidosis patient. Model system for revealing the intracellular transport of normal and mutated lysosomal enzymes.Biochem J. 1999 Jun 1;340 ( Pt 2)(Pt 2):467-74. doi: 10.1042/bj3400467. Biochem J. 1999. PMID: 10333491 Free PMC article.
-
Cathepsin A/protective protein: an unusual lysosomal multifunctional protein.Cell Mol Life Sci. 1999 Dec;56(11-12):894-907. doi: 10.1007/s000180050482. Cell Mol Life Sci. 1999. PMID: 11212324 Free PMC article. Review.
-
Molecular and biochemical analysis of protective protein/cathepsin A mutations: correlation with clinical severity in galactosialidosis.Hum Mol Genet. 1996 Dec;5(12):1977-87. doi: 10.1093/hmg/5.12.1977. Hum Mol Genet. 1996. PMID: 8968752
-
Lack of PPCA expression only partially coincides with lysosomal storage in galactosialidosis mice: indirect evidence for spatial requirement of the catalytic rather than the protective function of PPCA.Hum Mol Genet. 1998 Oct;7(11):1787-94. doi: 10.1093/hmg/7.11.1787. Hum Mol Genet. 1998. PMID: 9736781
-
The biochemistry and clinical features of galactosialidosis.Biochim Biophys Acta. 1994 Feb 22;1225(3):244-54. doi: 10.1016/0925-4439(94)90002-7. Biochim Biophys Acta. 1994. PMID: 8312369 Review.
Cited by
-
Proteolytic activation of human cathepsin A.J Biol Chem. 2014 Apr 25;289(17):11592-11600. doi: 10.1074/jbc.M113.524280. Epub 2014 Mar 5. J Biol Chem. 2014. PMID: 24599961 Free PMC article.
-
Stable expression of protective protein/cathepsin A-green fluorescent protein fusion genes in a fibroblastic cell line from a galactosialidosis patient. Model system for revealing the intracellular transport of normal and mutated lysosomal enzymes.Biochem J. 1999 Jun 1;340 ( Pt 2)(Pt 2):467-74. doi: 10.1042/bj3400467. Biochem J. 1999. PMID: 10333491 Free PMC article.
-
Galactosialidosis: review and analysis of CTSA gene mutations.Orphanet J Rare Dis. 2013 Aug 2;8:114. doi: 10.1186/1750-1172-8-114. Orphanet J Rare Dis. 2013. PMID: 23915561 Free PMC article. Review.
-
Galactosialidosis: historic aspects and overview of investigated and emerging treatment options.Expert Opin Orphan Drugs. 2017;5(2):131-141. doi: 10.1080/21678707.2016.1266933. Epub 2016 Dec 14. Expert Opin Orphan Drugs. 2017. PMID: 28603679 Free PMC article.
-
Next-generation sequencing of the whole mitochondrial genome identifies functionally deleterious mutations in patients with multiple sclerosis.PLoS One. 2022 Feb 7;17(2):e0263606. doi: 10.1371/journal.pone.0263606. eCollection 2022. PLoS One. 2022. PMID: 35130313 Free PMC article.
References
-
- d’Azzo A, Andria G, Strisciuglio P, Galjaard H. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver C R, Beaudet A L, Sly W S, Valle D, editors. New York: McGraw–Hill; 1995. pp. 2825–2837.
-
- Wenger D A, Tarby T J, Wharton C. Biochem Biophys Res Commun. 1978;82:589–595. - PubMed
-
- Andria G, Strisciuglio P, Pontarelli G, Sly W S, Dodson W E. In: Sialidases and Sialidosis. Perspectives in Inherited Metabolic Diseases. Tettamanti G, Durand P, Di Donato S, editors. Milano, Italy: Edizioni Ermes; 1981. pp. 379–395.
-
- Galjart N J, Gillemans N, Harris A, van der Horst G T J, Verheijen F W, Galjaard H, d’Azzo A. Cell. 1988;54:755–764. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
