

HFE gene knockout produces mouse model of hereditary hemochromatosis
- PMID: 9482913
- PMCID: PMC19387
- DOI: 10.1073/pnas.95.5.2492
HFE gene knockout produces mouse model of hereditary hemochromatosis
Abstract
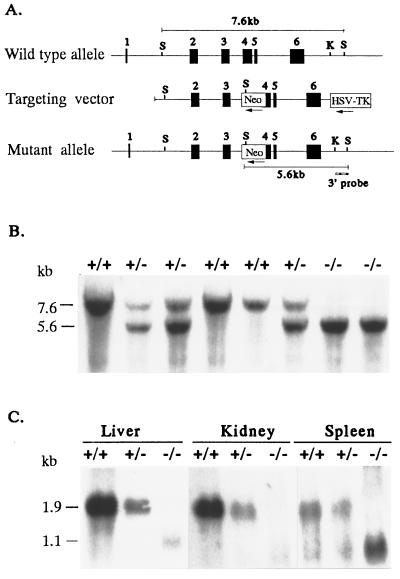
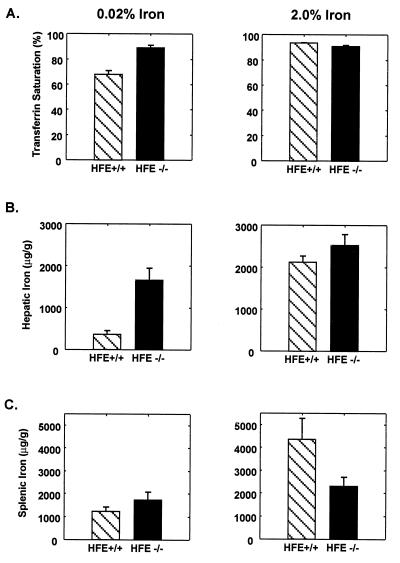
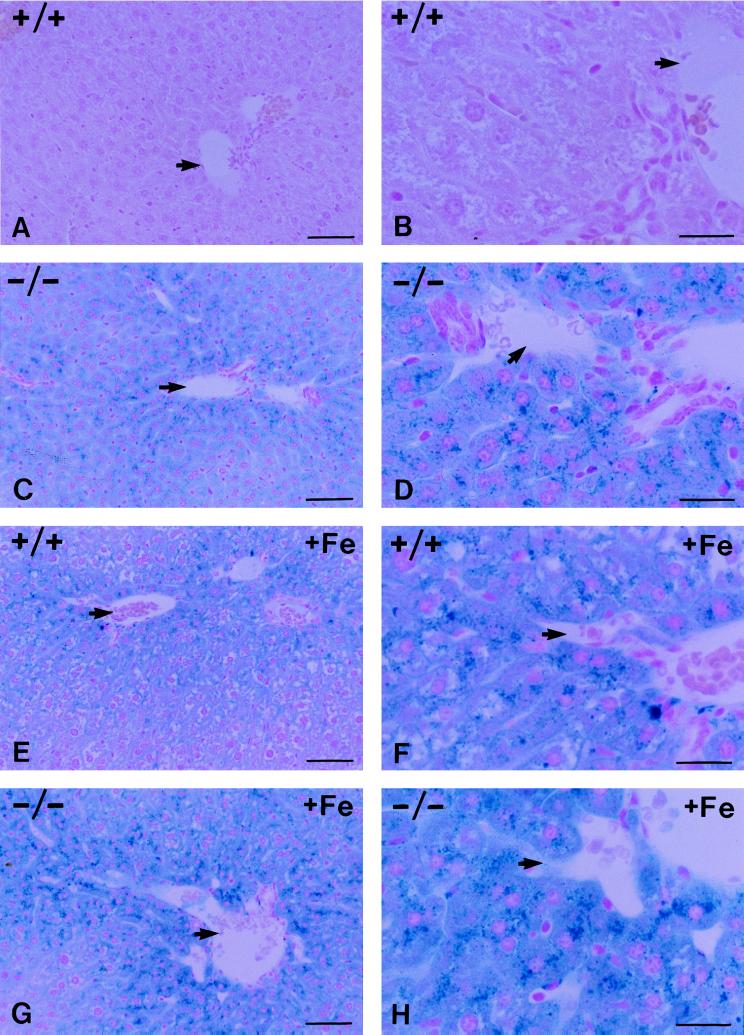
Hereditary hemochromatosis (HH) is a common autosomal recessive disease characterized by increased iron absorption and progressive iron storage that results in damage to major organs in the body. Recently, a candidate gene for HH called HFE encoding a major histocompatibility complex class I-like protein was identified by positional cloning. Nearly 90% of Caucasian HH patients have been found to be homozygous for the same mutation (C282Y) in the HFE gene. To test the hypothesis that the HFE gene is involved in regulation of iron homeostasis, we studied the effects of a targeted disruption of the murine homologue of the HFE gene. The HFE-deficient mice showed profound differences in parameters of iron homeostasis. Even on a standard diet, by 10 weeks of age, fasting transferrin saturation was significantly elevated compared with normal littermates (96 +/- 5% vs. 77 +/- 3%, P < 0.007), and hepatic iron concentration was 8-fold higher than that of wild-type littermates (2,071 +/- 450 vs. 255 +/- 23 microg/g dry wt, P < 0.002). Stainable hepatic iron in the HFE mutant mice was predominantly in hepatocytes in a periportal distribution. Iron concentrations in spleen, heart, and kidney were not significantly different. Erythroid parameters were normal, indicating that the anemia did not contribute to the increased iron storage. This study shows that the HFE protein is involved in the regulation of iron homeostasis and that mutations in this gene are responsible for HH. The knockout mouse model of HH will facilitate investigation into the pathogenesis of increased iron accumulation in HH and provide opportunities to evaluate therapeutic strategies for prevention or correction of iron overload.
Figures
Comment in
-
Targeted disruption of the HFE gene.Proc Natl Acad Sci U S A. 1998 Mar 3;95(5):2033-4. doi: 10.1073/pnas.95.5.2033. Proc Natl Acad Sci U S A. 1998. PMID: 9482831 Free PMC article. Review. No abstract available.
References
-
- Cartwright G E, Edwards C Q, Kravitz K, Skolnick M, Amos D B, Johnson A, Buskjaer L. N Engl J Med. 1979;301:175–179. - PubMed
-
- Borwein S T, Ghent C N, Flanagan P R, Chamberlain M J, Valberg L S. Clin Invest Med. 1983;6:171–179. - PubMed
-
- Edwards C Q, Griffen L M, Goldgar D, Drummond C, Skolnick M H, Kushner J P. N Engl J Med. 1988;318:1355–1362. - PubMed
-
- Bacon B R, Tavill A S. In: Hepatology: A Textbook of Liver Disease. Zakim D, Boyer T D, editors. Philadelphia: Saunders; 1996. pp. 1439–1472.
-
- Barton J C, Bertoli L F. Nat Med. 1996;2:394–395. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
