

Cytokine kinetics and other host factors in response to pneumococcal pulmonary infection in mice
- PMID: 9488375
- PMCID: PMC107995
- DOI: 10.1128/IAI.66.3.912-922.1998
Cytokine kinetics and other host factors in response to pneumococcal pulmonary infection in mice
Abstract
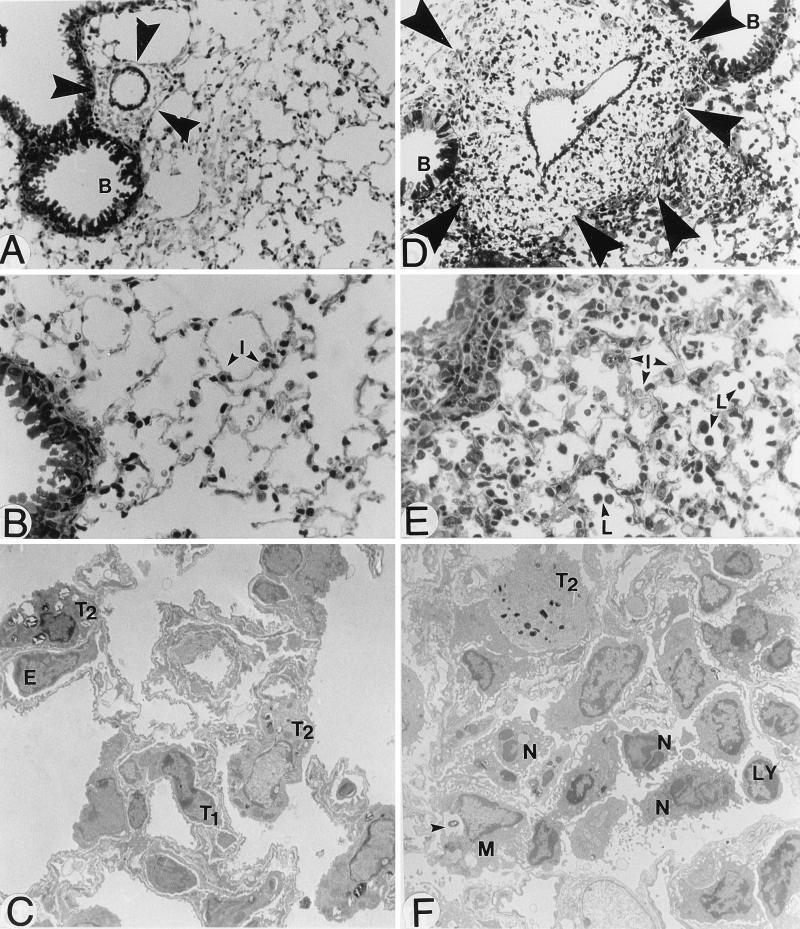
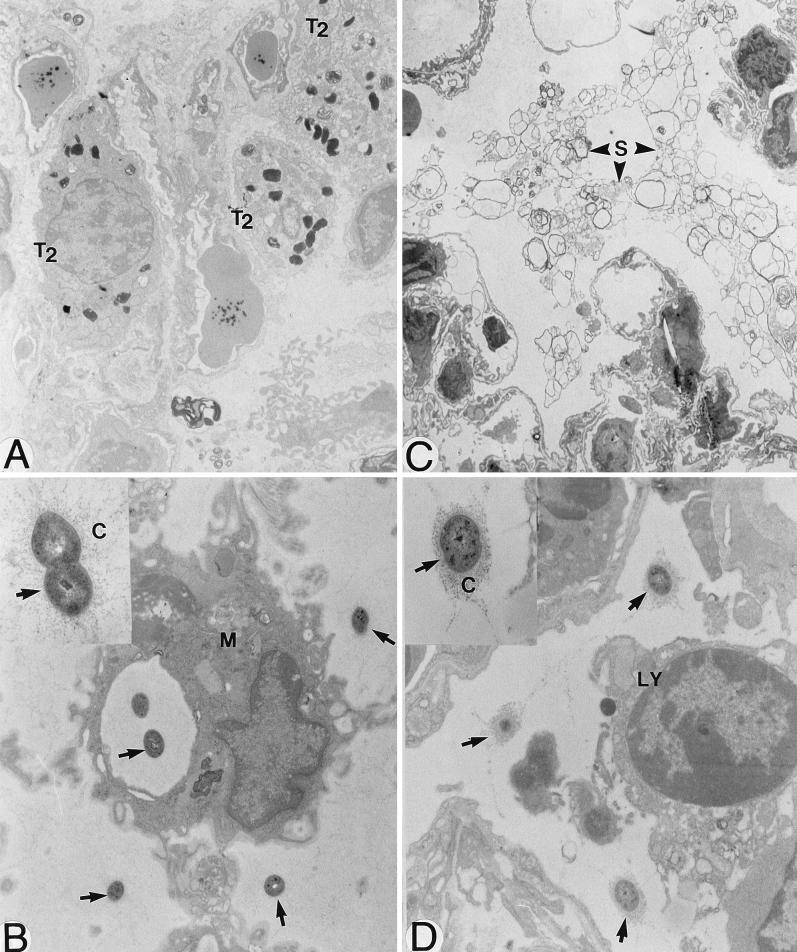
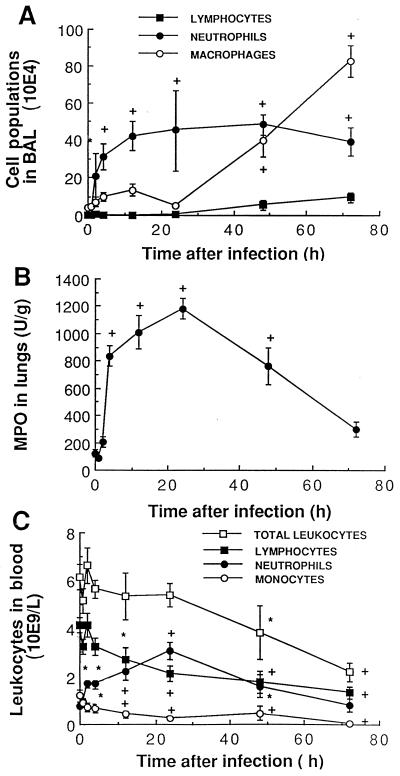
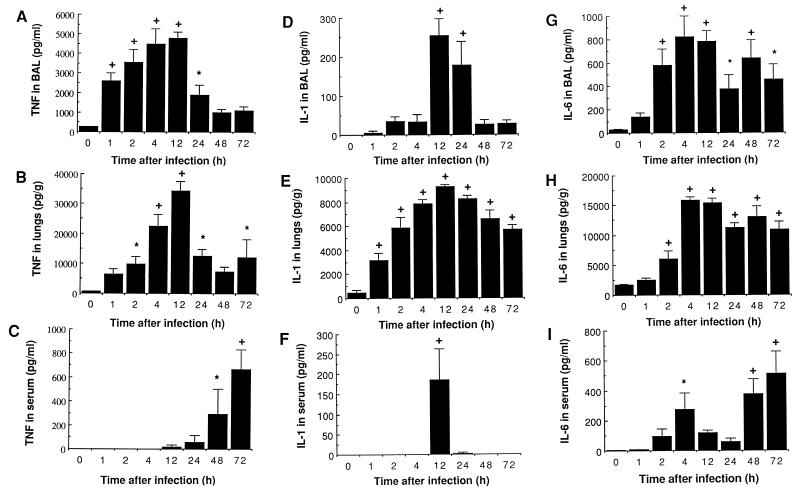
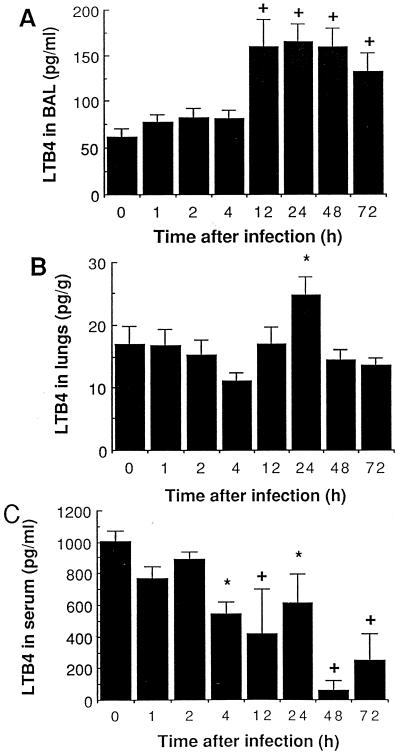
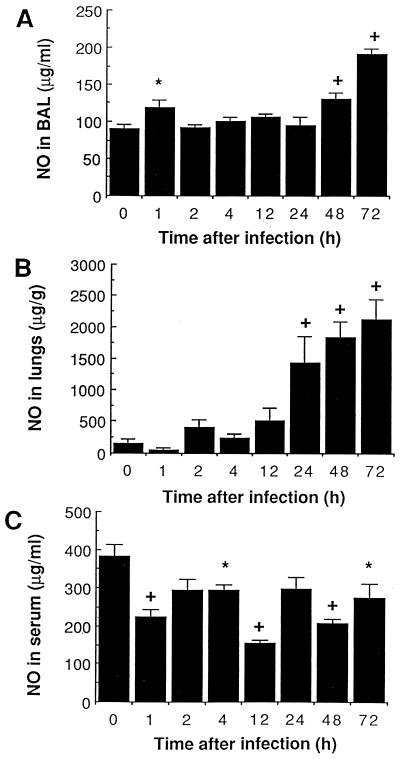
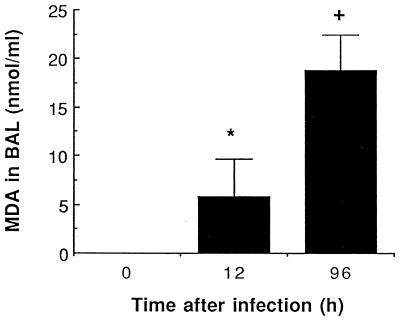
There is a need for more insight into the pathogenesis of Streptococcus pneumoniae pneumonia, as the fatality rate associated with this disease remains high despite appropriate antibiotherapy. The host response to pneumococci was investigated after intranasal inoculation of CD1 mice with 10(7) log-phase CFU of bacteria. We identified five major pathogenesis steps from initial infection to death. In step 1 (0 to 4 h), there was ineffective phagocytosis by alveolar macrophages, with concurrent release of tumor necrosis factor alpha (TNF), interleukin-6 (IL-6), and nitric oxide (NO) in bronchoalveolar lavage (BAL) fluid, TNF, IL-6, and interleukin-1 alpha (IL-1) in lung tissues, and IL-6 in serum, which were associated with tachypnea and hemoconcentration. In step 2 (4 to 24 h), bacterial growth in alveoli and polymorphonuclear cell recruitment from bloodstream to lung tissue (high myeloperoxidase levels) to alveoli were associated with high release of all three cytokines and leukotriene B4 (LTB4) in tissue and BAL fluid, as well as transient spillover of IL-1 in serum. In step 3 (24 to 48 h), despite downregulation of TNF and IL-1 in BAL fluid and lungs, there was appearance of injury to alveolar ultrastructure, edema to interstitium, and increase in lung weight as well as regeneration of type II pneumocytes and increased secretion of surfactant; bacteria progressed from alveoli to tissue to blood, and body weight loss occurred. In step 4 (48 to 72 h), strong monocyte recruitment from blood to alveoli was associated with high NO release in tissue and BAL fluid, but there was also noticeable lymphocyte recruitment and leukopenia; bacteremia was associated with TNF and IL-6 release in blood and thrombocytopenia. In step 5 (72 to 96 h), severe airspace disorganization, lipid peroxidation (high malondialdehyde release in BAL fluid), and diffuse tissue damage coincided with high NO levels; there was further increase in lung weight and bacterial growth, loss in body weight, and high mortality rate. Delineation of the sequential steps that contribute to the pathogenesis of pneumococcal pneumonia may generate markers of evolution of disease and lead to better targeted intervention.
Figures
References
-
- Abbas A K, Lichtman A H, Pober J S. Effector mechanisms of immune responses. Cytokines. In: Wonsiewicz M J, editor. Cellular and molecular immunology. W. B. Philadelphia, Pa: Saunders Company; 1991. pp. 232–235.
-
- Aihara M, Nakazawa T, Dobashi K, Joshita T, Kojima M, Onai M, Mori M. A selective pulmonary thrombosis associated with sepsis-induced disseminated intravascular coagulation. Intern Med. 1997;36:97–101. - PubMed
-
- Amura C R, Fontan P A, Sanjuan N, Sordelli D O. The effect of treatment with interleukin-1 and tumor necrosis factor on Pseudomonas aeruginosa lung infection in a granulocytopenic mouse model. Clin Immunol Immunopathol. 1994;73:261–266. - PubMed
-
- Anderson, V. M., and T. Turner. 1991. Histopathology of childhood pneumonia in developing countries. Rev. Infect. Dis. 13(Suppl. 6):S470–S476. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
