

Mapping viral DNA specificity to the central region of integrase by using functional human immunodeficiency virus type 1/visna virus chimeric proteins
- PMID: 9499023
- PMCID: PMC109462
- DOI: 10.1128/JVI.72.3.1744-1753.1998
Mapping viral DNA specificity to the central region of integrase by using functional human immunodeficiency virus type 1/visna virus chimeric proteins
Abstract
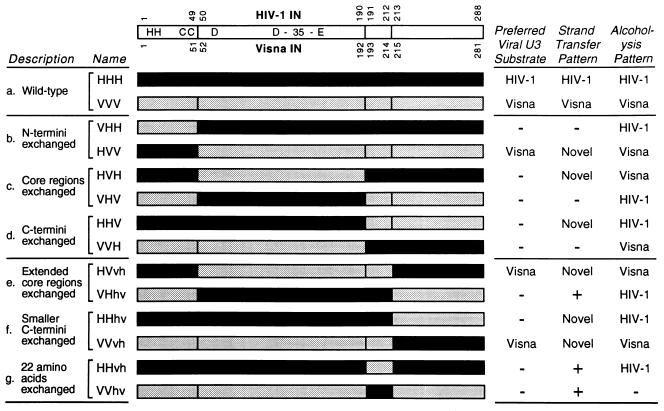
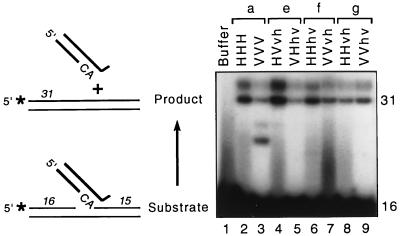
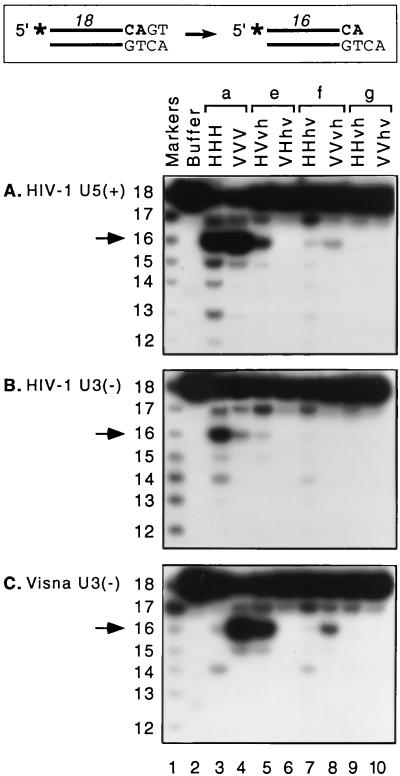
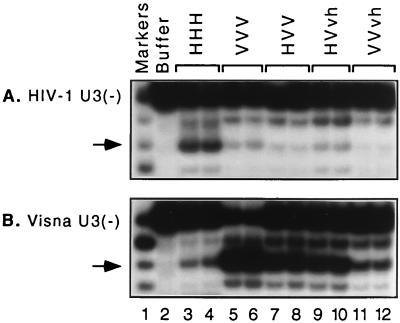
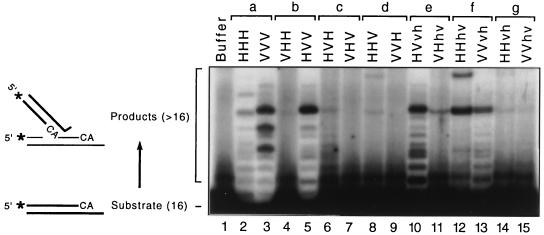
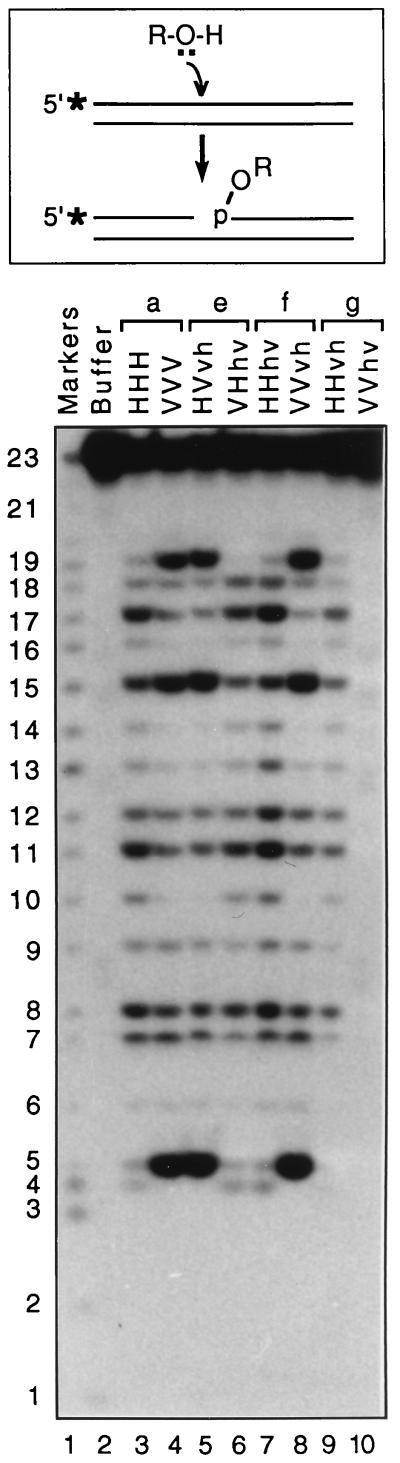
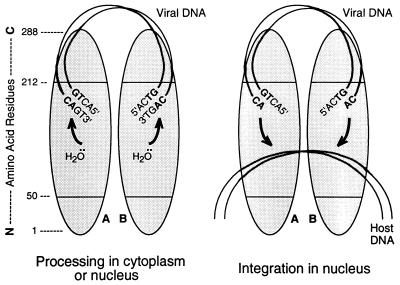
We previously described the construction and analysis of the first set of functional chimeric lentivirus integrases, involving exchange of the N-terminal, central, and C-terminal regions of the human immunodeficiency virus type 1 (HIV-1) and visna virus integrase (IN) proteins. Based on those results, additional HIV-1/visna virus chimeric integrases were designed and purified. Each of the chimeric enzymes was functional in at least one oligonucleotide-based IN assay. Of a total of 12 chimeric IN proteins, 3 exhibit specific viral DNA processing, 9 catalyze insertion of viral DNA ends, 12 can reverse that reaction, and 11 are active for nonspecific alcoholysis. Functional data obtained with the processing assay indicate that the central region of the protein is responsible for viral DNA specificity. Target site selection for nonspecific alcoholysis again mapped to the central domain of IN, confirming our previous data indicating that this region can position nonviral DNA for nucleophilic attack. However, the chimeric proteins created patterns of viral DNA insertion distinct from that of either wild-type IN, suggesting that interactions between regions of IN influence target site selection for viral DNA integration. The results support a new model for the functional organization of IN in which viral DNA initially binds nonspecifically to the C-terminal portion of IN but the catalytic central region of the enzyme has a prominent role both in specific recognition of viral DNA ends and in positioning the host DNA for viral DNA integration.
Figures
Similar articles
-
Mapping domains of retroviral integrase responsible for viral DNA specificity and target site selection by analysis of chimeras between human immunodeficiency virus type 1 and visna virus integrases.J Virol. 1995 Sep;69(9):5687-96. doi: 10.1128/JVI.69.9.5687-5696.1995. J Virol. 1995. PMID: 7637015 Free PMC article.
-
Mapping target site selection for the non-specific nuclease activities of retroviral integrase.Virus Res. 2000 Jan;66(1):87-100. doi: 10.1016/s0168-1702(99)00126-4. Virus Res. 2000. PMID: 10653920
-
Characterization of chimeric enzymes between caprine arthritis--encephalitis virus, maedi--visna virus and human immunodeficiency virus type 1 integrases expressed in Escherichia coli.J Gen Virol. 2001 Jan;82(Pt 1):139-148. doi: 10.1099/0022-1317-82-1-139. J Gen Virol. 2001. PMID: 11125167
-
Substrate recognition by retroviral integrases.Adv Virus Res. 1999;52:371-95. doi: 10.1016/s0065-3527(08)60307-3. Adv Virus Res. 1999. PMID: 10384243 Review.
-
Site-specific integration of retroviral DNA in human cells using fusion proteins consisting of human immunodeficiency virus type 1 integrase and the designed polydactyl zinc-finger protein E2C.Methods. 2009 Apr;47(4):269-76. doi: 10.1016/j.ymeth.2009.01.001. Epub 2009 Jan 30. Methods. 2009. PMID: 19186211 Free PMC article. Review.
Cited by
-
Symmetrical recognition of cellular DNA target sequences during retroviral integration.Proc Natl Acad Sci U S A. 2005 Apr 26;102(17):5903-4. doi: 10.1073/pnas.0502045102. Epub 2005 Apr 19. Proc Natl Acad Sci U S A. 2005. PMID: 15840713 Free PMC article. No abstract available.
-
Retroviral DNA integration.Microbiol Mol Biol Rev. 1999 Dec;63(4):836-43, table of contents. doi: 10.1128/MMBR.63.4.836-843.1999. Microbiol Mol Biol Rev. 1999. PMID: 10585967 Free PMC article. Review.
-
Dissecting the role of the N-terminal domain of human immunodeficiency virus integrase by trans-complementation analysis.J Virol. 1999 Apr;73(4):3176-83. doi: 10.1128/JVI.73.4.3176-3183.1999. J Virol. 1999. PMID: 10074170 Free PMC article.
-
Division of labor within human immunodeficiency virus integrase complexes: determinants of catalysis and target DNA capture.J Virol. 2005 Dec;79(24):15376-87. doi: 10.1128/JVI.79.24.15376-15387.2005. J Virol. 2005. PMID: 16306609 Free PMC article.
-
Mechanisms of LTR-Retroelement Transposition: Lessons from Drosophila melanogaster.Viruses. 2017 Apr 16;9(4):81. doi: 10.3390/v9040081. Viruses. 2017. PMID: 28420154 Free PMC article. Review.
References
-
- Andrake M D, Skalka A M. Multimerization determinants reside in both the catalytic core and C terminus of avian sarcoma virus integrase. J Biol Chem. 1995;270:29299–29306. - PubMed
-
- Andrake M D, Skalka A M. Retroviral integrase, putting the pieces together. J Biol Chem. 1996;271:19633–19636. - PubMed
-
- Barsov E V, Huber W E, Marcotrigiano J, Clark P K, Clark A D, Arnold E, Hughes S H. Inhibition of human immunodeficiency virus type 1 integrase by the Fab fragment of a specific monoclonal antibody suggests that different multimerization states are required for different enzymatic functions. J Virol. 1996;70:4484–4494. - PMC - PubMed
-
- Bizub-Bender D, Kulkosky J, Skalka A M. Monoclonal antibodies against HIV type 1 integrase: clues to molecular structure. AIDS Res Hum Retroviruses. 1994;10:1105–1115. - PubMed
-
- Bujacz G, Jaskolski M, Alexandratos J, Wlodawer A, Merkel G, Katz R A, Skalka A M. High-resolution structure of the catalytic domain of avian sarcoma virus integrase. J Mol Biol. 1995;253:333–346. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
