

Structure-based prediction of the stability of transmembrane helix-helix interactions: the sequence dependence of glycophorin A dimerization
- PMID: 9520409
- PMCID: PMC19879
- DOI: 10.1073/pnas.95.7.3583
Structure-based prediction of the stability of transmembrane helix-helix interactions: the sequence dependence of glycophorin A dimerization
Abstract
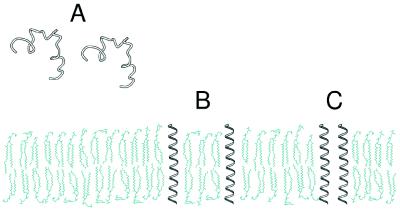
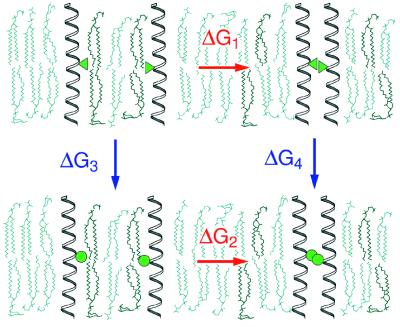
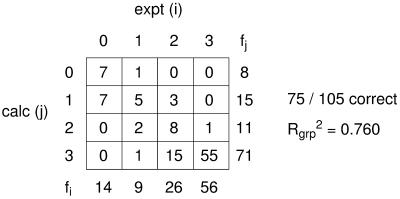
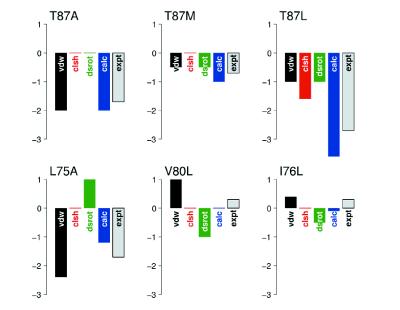
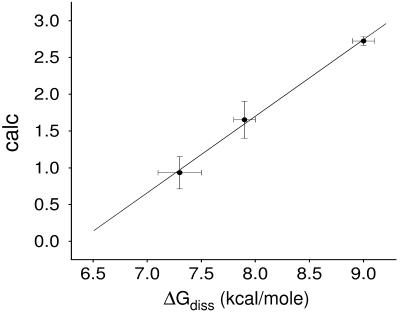
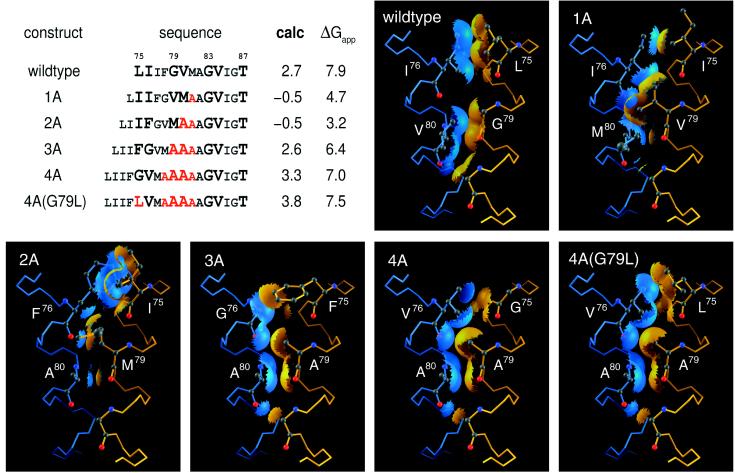
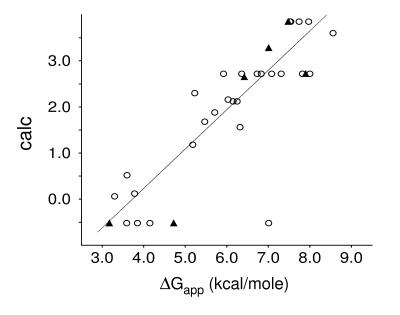
The ability to predict the effects of point mutations on the interaction of alpha-helices within membranes would represent a significant step toward understanding the folding and stability of membrane proteins. We use structure-based empirical parameters representing steric clashes, favorable van der Waals interactions, and restrictions of side-chain rotamer freedom to explain the relative dimerization propensities of 105 hydrophobic single-point mutants of the glycophorin A (GpA) transmembrane domain. Although the structure at the dimer interface is critical to our model, changes in side-chain hydrophobicity are uncorrelated with dimer stability, indicating that the hydrophobic effect does not influence transmembrane helix-helix association. Our model provides insights into the compensatory effects of multiple mutations and shows that helix-helix interactions dominate the formation of specific structures.
Figures
References
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
