

Human PEX1 cloned by functional complementation on a CHO cell mutant is responsible for peroxisome-deficient Zellweger syndrome of complementation group I
- PMID: 9539740
- PMCID: PMC22492
- DOI: 10.1073/pnas.95.8.4350
Human PEX1 cloned by functional complementation on a CHO cell mutant is responsible for peroxisome-deficient Zellweger syndrome of complementation group I
Abstract
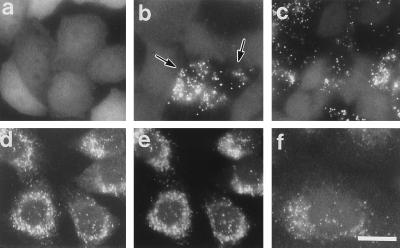
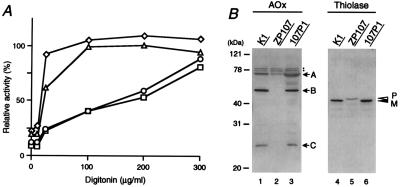
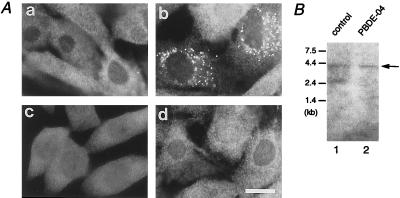
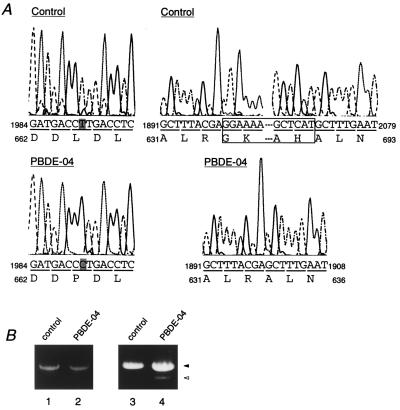
The peroxisome biogenesis disorders (PBDs), including Zellweger syndrome (ZS) and neonatal adrenoleukodystrophy (NALD), are autosomal recessive diseases caused by defects in peroxisome assembly, for which at least 10 complementation groups have been reported. We have isolated a human PEX1 cDNA (HsPEX1) by functional complementation of peroxisome deficiency of a mutant Chinese hamster ovary (CHO) cell line, ZP107, transformed with peroxisome targeting signal type 1-tagged "enhanced" green fluorescent protein. This cDNA encodes a hydrophilic protein (Pex1p) comprising 1,283 amino acids, with high homology to the AAA-type ATPase family. A stable transformant of ZP107 with HsPEX1 was morphologically and biochemically restored for peroxisome biogenesis. HsPEX1 expression restored peroxisomal protein import in fibroblasts from three patients with ZS and NALD of complementation group I (CG-I), which is the highest-incidence PBD. A CG-I ZS patient (PBDE-04) possessed compound heterozygous, inactivating mutations: a missense point mutation resulting in Leu-664 --> Pro and a deletion of the sequence from Gly-634 to His-690 presumably caused by missplicing (splice site mutation). Both PBDE-04 PEX1 cDNAs were defective in peroxisome-restoring activity when expressed in the patient fibroblasts as well as in ZP107 cells. These results demonstrate that PEX1 is the causative gene for CG-I peroxisomal disorders.
Figures

Similar articles
-
Phenotype-genotype relationships in peroxisome biogenesis disorders of PEX1-defective complementation group 1 are defined by Pex1p-Pex6p interaction.Biochem J. 2001 Jul 15;357(Pt 2):417-26. doi: 10.1042/0264-6021:3570417. Biochem J. 2001. PMID: 11439091 Free PMC article.
-
Human PEX19: cDNA cloning by functional complementation, mutation analysis in a patient with Zellweger syndrome, and potential role in peroxisomal membrane assembly.Proc Natl Acad Sci U S A. 1999 Mar 2;96(5):2116-21. doi: 10.1073/pnas.96.5.2116. Proc Natl Acad Sci U S A. 1999. PMID: 10051604 Free PMC article.
-
Human PEX1 is mutated in complementation group 1 of the peroxisome biogenesis disorders.Nat Genet. 1997 Dec;17(4):449-52. doi: 10.1038/ng1297-449. Nat Genet. 1997. PMID: 9398848
-
[Human peroxisome-deficient disorders and pathogenic gene].Rinsho Shinkeigaku. 1994 Dec;34(12):1219-21. Rinsho Shinkeigaku. 1994. PMID: 7539728 Review. Japanese.
-
[Molecular biology of peroxisome biogenesis].Nihon Rinsho. 1993 Sep;51(9):2336-42. Nihon Rinsho. 1993. PMID: 8411711 Review. Japanese.
Cited by
-
PEX3 is the causal gene responsible for peroxisome membrane assembly-defective Zellweger syndrome of complementation group G.Am J Hum Genet. 2000 Oct;67(4):976-81. doi: 10.1086/303086. Epub 2000 Aug 31. Am J Hum Genet. 2000. PMID: 10968777 Free PMC article.
-
Characterization of two common 5' polymorphisms in PEX1 and correlation to survival in PEX1 peroxisome biogenesis disorder patients.BMC Med Genet. 2011 Aug 16;12:109. doi: 10.1186/1471-2350-12-109. BMC Med Genet. 2011. PMID: 21846392 Free PMC article.
-
Fusion of small peroxisomal vesicles in vitro reconstructs an early step in the in vivo multistep peroxisome assembly pathway of Yarrowia lipolytica.J Cell Biol. 2000 Jan 10;148(1):29-44. doi: 10.1083/jcb.148.1.29. J Cell Biol. 2000. PMID: 10629216 Free PMC article.
-
Shuttling mechanism of peroxisome targeting signal type 1 receptor Pex5: ATP-independent import and ATP-dependent export.Mol Cell Biol. 2005 Dec;25(24):10822-32. doi: 10.1128/MCB.25.24.10822-10832.2005. Mol Cell Biol. 2005. PMID: 16314507 Free PMC article.
-
Crystal structure of the conserved N-terminal domain of the peroxisomal matrix protein import receptor, Pex14p.Proc Natl Acad Sci U S A. 2009 Jan 13;106(2):417-21. doi: 10.1073/pnas.0808681106. Epub 2009 Jan 2. Proc Natl Acad Sci U S A. 2009. PMID: 19122147 Free PMC article.
References
-
- van den Bosch H, Schutgens R B H, Wanders R J A, Tager J M. Annu Rev Biochem. 1992;61:157–197. - PubMed
-
- Lazarow P B, Fujiki Y. Annu Rev Cell Biol. 1985;1:489–530. - PubMed
-
- Lazarow P B, Moser H W. In: The Metabolic Basis of Inherited Disease. 7th Ed. Scriver C R, Beaudet A I, Sly W S, Valle D, editors. New York: McGraw-Hill; 1995. pp. 2287–2324.
-
- Fujiki Y. Biochim Biophys Acta. 1997;1361:235–250. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
