

Architectural specificity in chromatin structure at the TATA box in vivo: nucleosome displacement upon beta-phaseolin gene activation
- PMID: 9539814
- PMCID: PMC22566
- DOI: 10.1073/pnas.95.8.4772
Architectural specificity in chromatin structure at the TATA box in vivo: nucleosome displacement upon beta-phaseolin gene activation
Abstract
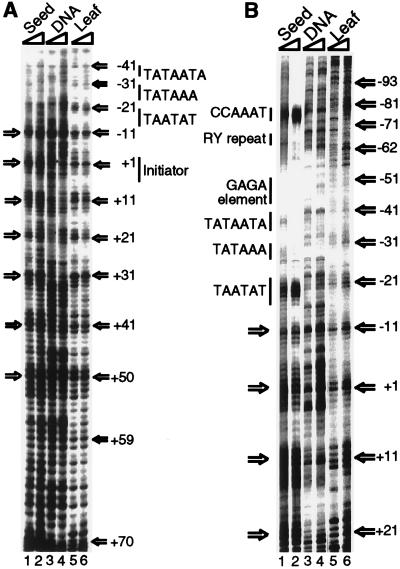
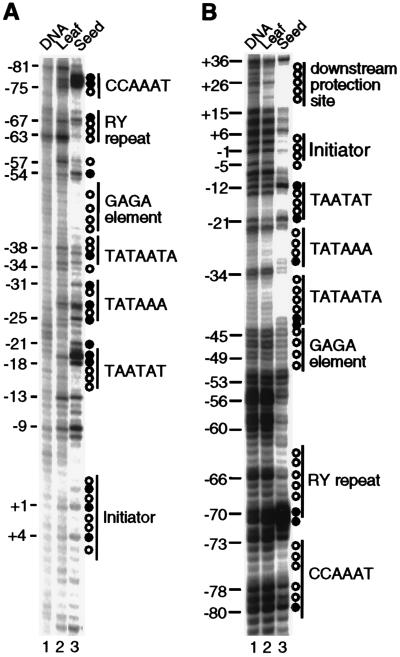
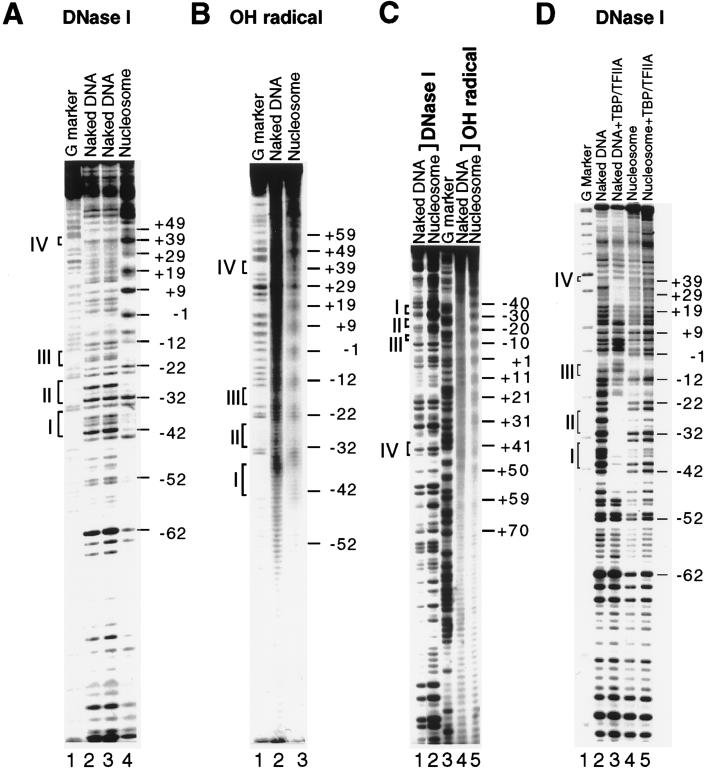
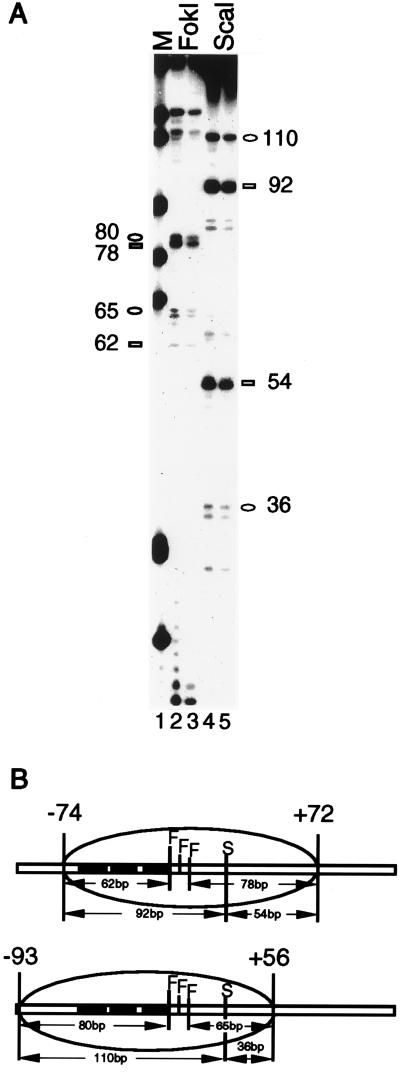
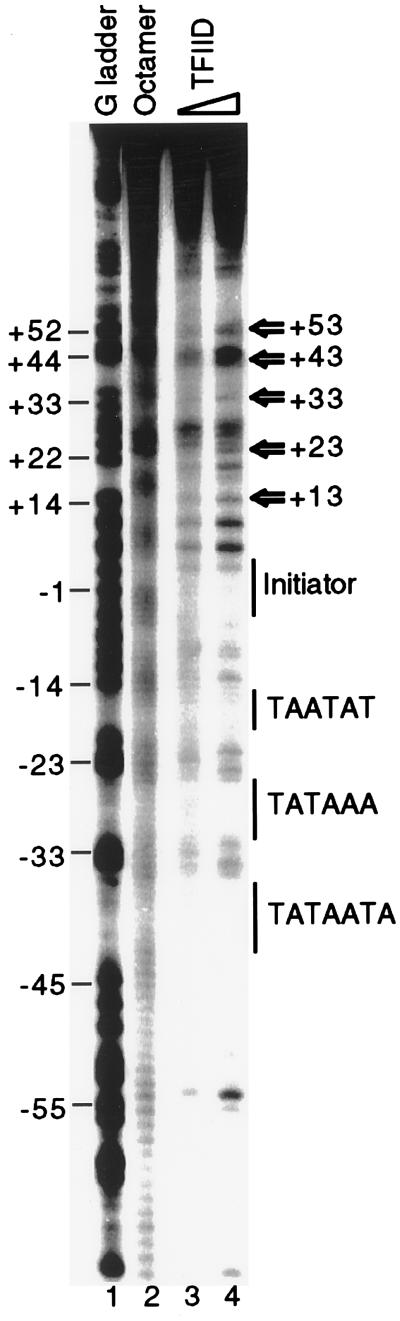
Extensive studies of the beta-phaseolin (phas) gene in transgenic tobacco have shown that it is highly active during seed embryogenesis but is completely silent in leaf and other vegetative tissues. In vivo footprinting revealed that the lack of even basal transcriptional activity in vegetative tissues is associated with the presence of a nucleosome that is rotationally positioned with base pair precision over three phased TATA boxes present in the phas promoter. Positioning is sequence-dependent because an identical rotational setting is obtained upon nucleosome reconstitution in vitro. A comparison of DNase I and dimethyl sulfate footprints in vivo and in vitro strongly suggests that this repressive chromatin architecture is remodeled concomitant with gene activation in the developing seed. This leads to the disruption of histone-mediated DNA wrapping and the assembly of the TATA boxes into a transcriptionally competent nucleoprotein complex.
Figures

References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
