

The permeability transition pore complex: a target for apoptosis regulation by caspases and bcl-2-related proteins
- PMID: 9547337
- PMCID: PMC2212234
- DOI: 10.1084/jem.187.8.1261
The permeability transition pore complex: a target for apoptosis regulation by caspases and bcl-2-related proteins
Abstract
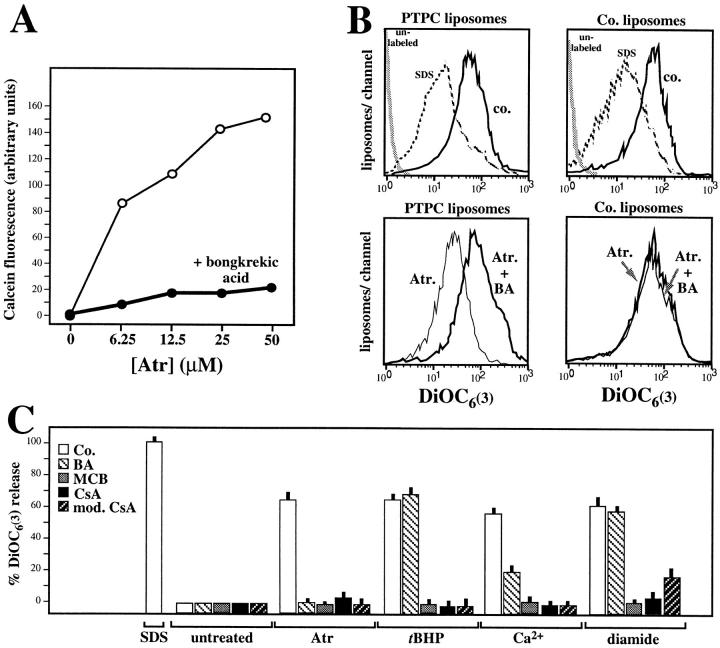
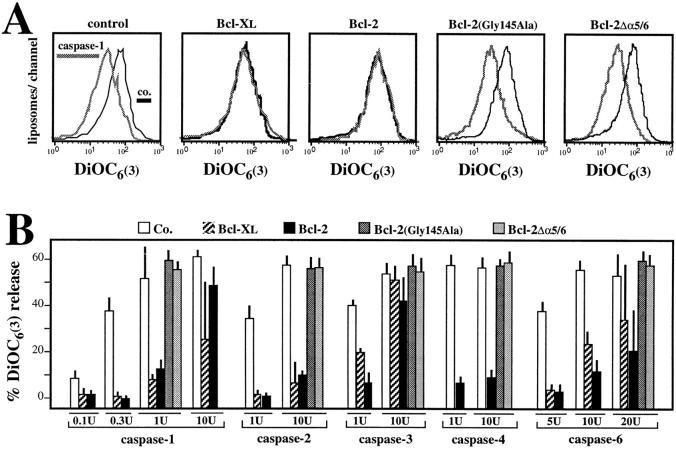
Early in programmed cell death (apoptosis), mitochondrial membrane permeability increases. This is at least in part due to opening of the permeability transition (PT) pore, a multiprotein complex built up at the contact site between the inner and the outer mitochondrial membranes. The PT pore has been previously implicated in clinically relevant massive cell death induced by toxins, anoxia, reactive oxygen species, and calcium overload. Here we show that PT pore complexes reconstituted in liposomes exhibit a functional behavior comparable with that of the natural PT pore present in intact mitochondria. The PT pore complex is regulated by thiol-reactive agents, calcium, cyclophilin D ligands (cyclosporin A and a nonimmunosuppressive cyclosporin A derivative), ligands of the adenine nucleotide translocator, apoptosis-related endoproteases (caspases), and Bcl-2-like proteins. Although calcium, prooxidants, and several recombinant caspases (caspases 1, 2, 3, 4, and 6) enhance the permeability of PT pore-containing liposomes, recombinant Bcl-2 or Bcl-XL augment the resistance of the reconstituted PT pore complex to pore opening. Mutated Bcl-2 proteins that have lost their cytoprotective potential also lose their PT modulatory capacity. In conclusion, the PT pore complex may constitute a crossroad of apoptosis regulation by caspases and members of the Bcl-2 family.
Figures




References
-
- Kroemer G, Zamzami N, Susin SA. Mitochondrial control of apoptosis. Immunol Today. 1997;18:44–51. - PubMed
-
- Kroemer G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med. 1997;3:614–620. - PubMed
-
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. . Cell. 1996;86:147–157. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
