

Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities
- PMID: 9560283
- PMCID: PMC20268
- DOI: 10.1073/pnas.95.9.5372
Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities
Abstract
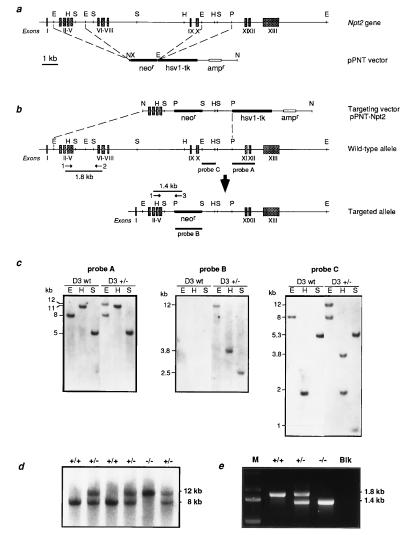
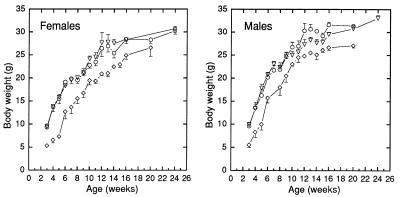
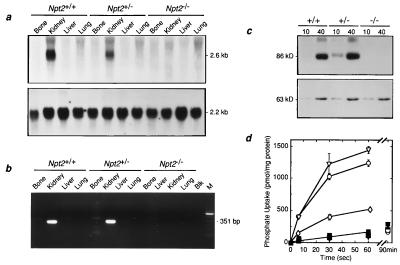
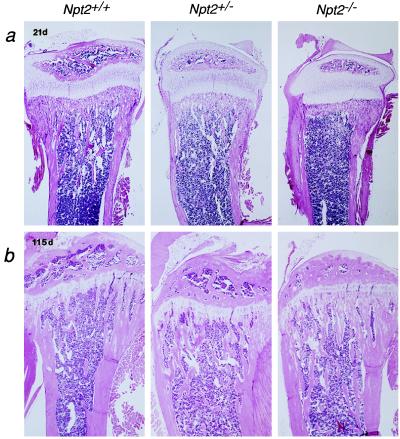
Npt2 encodes a renal-specific, brush-border membrane Na+-phosphate (Pi) cotransporter that is expressed in the proximal tubule where the bulk of filtered Pi is reabsorbed. Mice deficient in the Npt2 gene were generated by targeted mutagenesis to define the role of Npt2 in the overall maintenance of Pi homeostasis, determine its impact on skeletal development, and clarify its relationship to autosomal disorders of renal Pi reabsorption in humans. Homozygous mutants (Npt2(-/-)) exhibit increased urinary Pi excretion, hypophosphatemia, an appropriate elevation in the serum concentration of 1,25-dihydroxyvitamin D with attendant hypercalcemia, hypercalciuria and decreased serum parathyroid hormone levels, and increased serum alkaline phosphatase activity. These biochemical features are typical of patients with hereditary hypophosphatemic rickets with hypercalciuria (HHRH), a Mendelian disorder of renal Pi reabsorption. However, unlike HHRH patients, Npt2(-/-) mice do not have rickets or osteomalacia. At weaning, Npt2(-/-) mice have poorly developed trabecular bone and retarded secondary ossification, but, with increasing age, there is a dramatic reversal and eventual overcompensation of the skeletal phenotype. Our findings demonstrate that Npt2 is a major regulator of Pi homeostasis and necessary for normal skeletal development.
Figures
References
-
- Mizgala C L, Quamme G A. Physiol Rev. 1985;65:431–466. - PubMed
-
- Murer H, Werner A, Reshkin S, Wuarin R, Biber J. Am J Physiol. 1991;260:C885–C899. - PubMed
-
- Berndt T J, Knox F G. In: The Kidney: Physiology and Pathophysiology. Seldin D W, Giebisch G, editors. New York: Raven; 1992. pp. 2511–2532.
-
- Walker J J, Yan T S, Quamme G A. Am J Physiol. 1987;252:F226–F231. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
