

Multiple Grb2-mediated integrin-stimulated signaling pathways to ERK2/mitogen-activated protein kinase: summation of both c-Src- and focal adhesion kinase-initiated tyrosine phosphorylation events
- PMID: 9566877
- PMCID: PMC110637
- DOI: 10.1128/MCB.18.5.2571
Multiple Grb2-mediated integrin-stimulated signaling pathways to ERK2/mitogen-activated protein kinase: summation of both c-Src- and focal adhesion kinase-initiated tyrosine phosphorylation events
Abstract
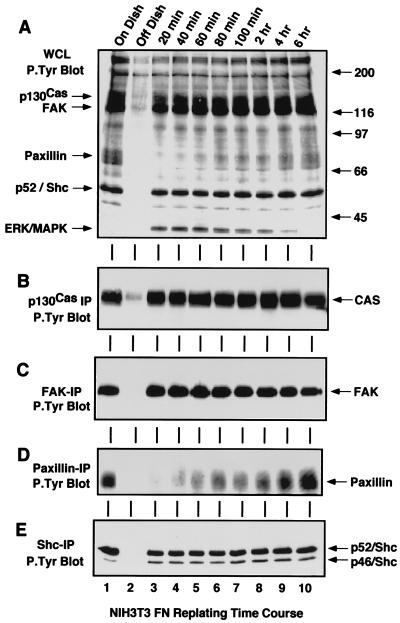
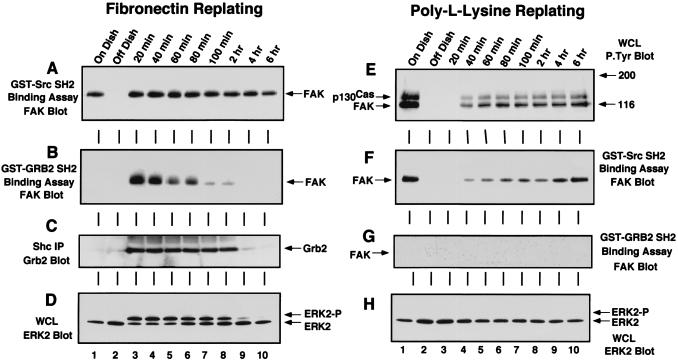
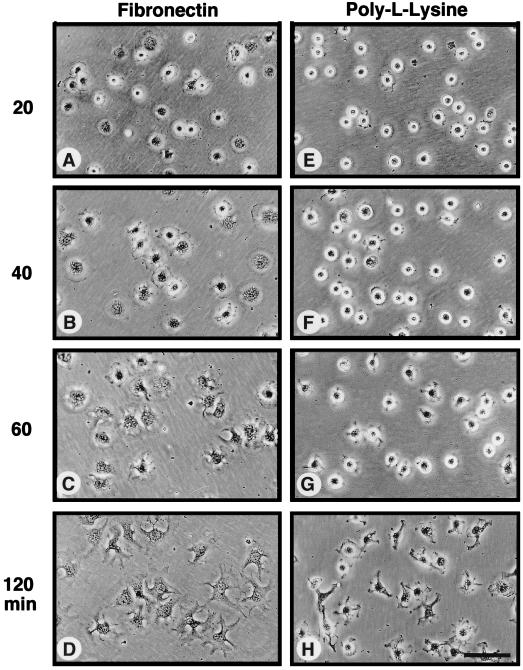
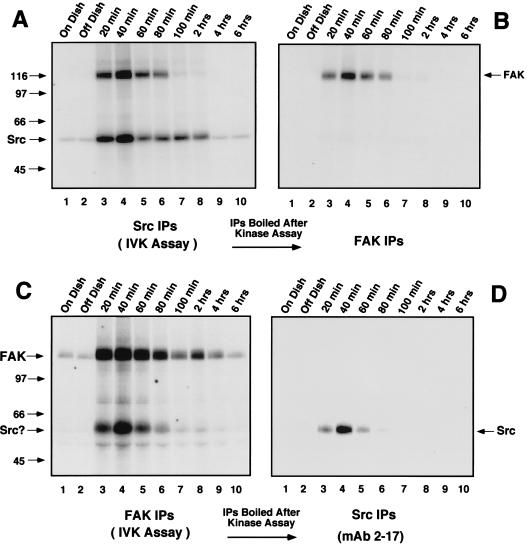
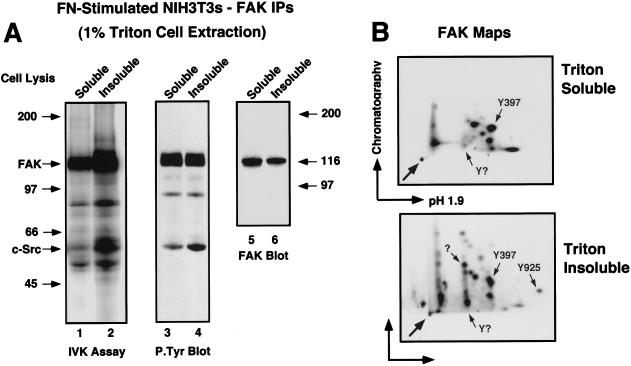
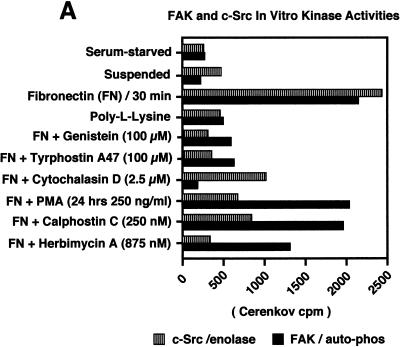
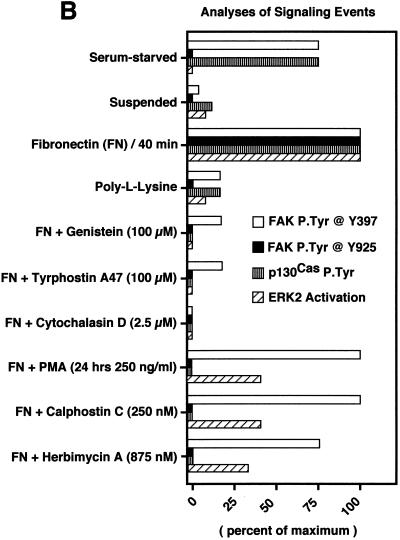
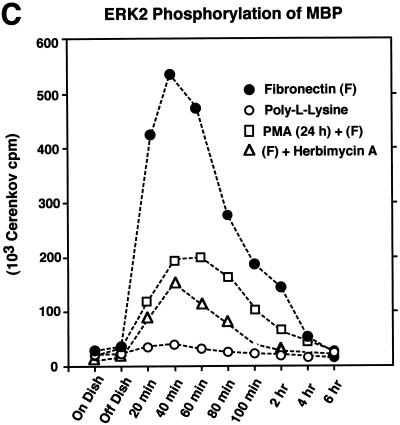
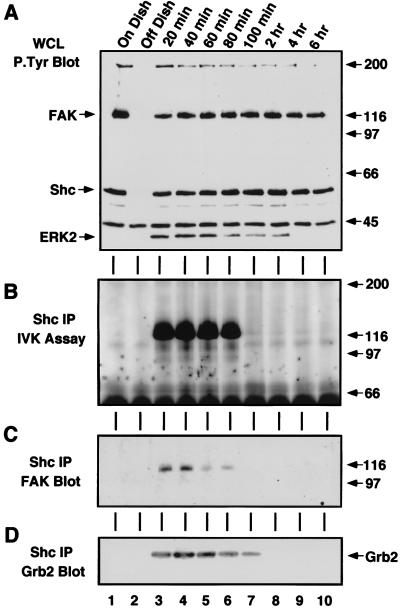
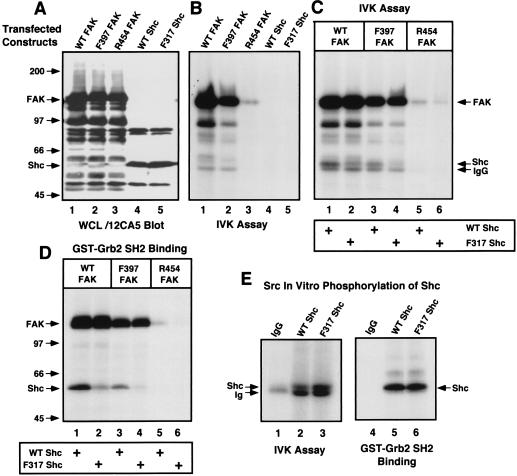
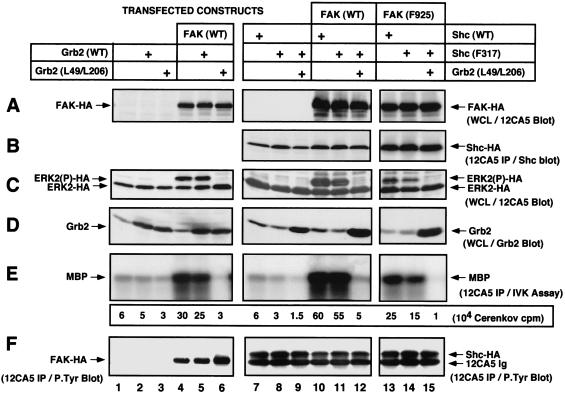
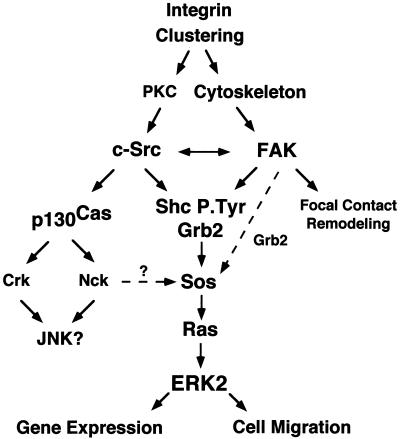
Fibronectin receptor integrin-mediated cell adhesion triggers intracellular signaling events such as the activation of the Ras/mitogen-activated protein (MAP) kinase cascade. In this study, we show that the nonreceptor protein-tyrosine kinases (PTKs) c-Src and focal adhesion kinase (FAK) can be independently activated after fibronectin (FN) stimulation and that their combined activity promotes signaling to extracellular signal-regulated kinase 2 (ERK2)/MAP kinase through multiple pathways upstream of Ras. FN stimulation of NIH 3T3 fibroblasts promotes c-Src and FAK association in the Triton-insoluble cell fraction, and the time course of FN-stimulated ERK2 activation paralleled that of Grb2 binding to FAK at Tyr-925 and Grb2 binding to Shc. Cytochalasin D treatment of fibroblasts inhibited FN-induced FAK in vitro kinase activity and signaling to ERK2, but it only partially inhibited c-Src activation. Treatment of fibroblasts with protein kinase C inhibitors or with the PTK inhibitor herbimycin A or PP1 resulted in reduced Src PTK activity, no Grb2 binding to FAK, and lowered levels of ERK2 activation. FN-stimulated FAK PTK activity was not significantly affected by herbimycin A treatment and, under these conditions, FAK autophosphorylation promoted Shc binding to FAK. In vitro, FAK directly phosphorylated Shc Tyr-317 to promote Grb2 binding, and in vivo Grb2 binding to Shc was observed in herbimycin A-treated fibroblasts after FN stimulation. Interestingly, c-Src in vitro phosphorylation of Shc promoted Grb2 binding to both wild-type and Phe-317 Shc. In vivo, Phe-317 Shc was tyrosine phosphorylated after FN stimulation of human 293T cells and its expression did not inhibit signaling to ERK2. Surprisingly, expression of Phe-925 FAK with Phe-317 Shc also did not block signaling to ERK2, whereas FN-stimulated signaling to ERK2 was inhibited by coexpression of an SH3 domain-inactivated mutant of Grb2. Our studies show that FN receptor integrin signaling upstream of Ras and ERK2 does not follow a linear pathway but that, instead, multiple Grb2-mediated interactions with Shc, FAK, and perhaps other yet-to-be-determined phosphorylated targets represent parallel signaling pathways that cooperate to promote maximal ERK2 activation.
Figures
References
-
- Blaikie P A, Fournier E, Dilworth S M, Birnbaum D, Borg J P, Margolis B. The role of the Shc phosphotyrosine interaction/phosphotyrosine binding domain and tyrosine phosphorylation sites in polyoma middle T antigen-mediated cell transformation. J Biol Chem. 1997;272:20671–20677. - PubMed
-
- Boyle W J, van der Geer P, Hunter T. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol. 1991;201:110–149. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous