

Commitment and effector phases of the physiological cell death pathway elucidated with respect to Bcl-2 caspase, and cyclin-dependent kinase activities
- PMID: 9566910
- PMCID: PMC110670
- DOI: 10.1128/MCB.18.5.2912
Commitment and effector phases of the physiological cell death pathway elucidated with respect to Bcl-2 caspase, and cyclin-dependent kinase activities
Abstract
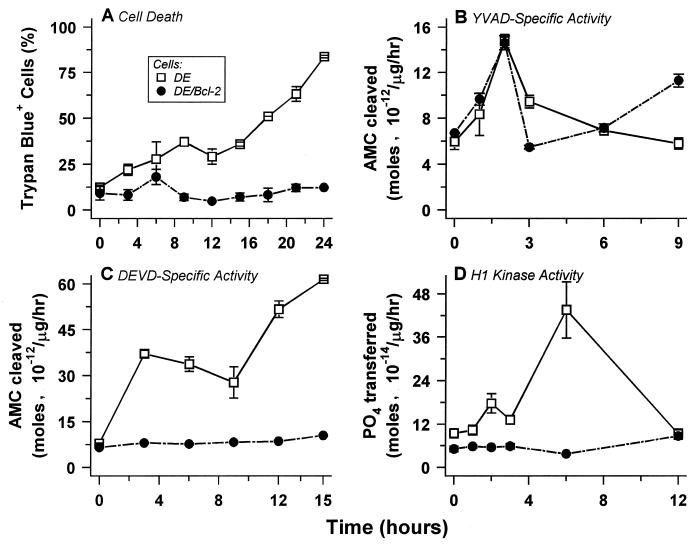
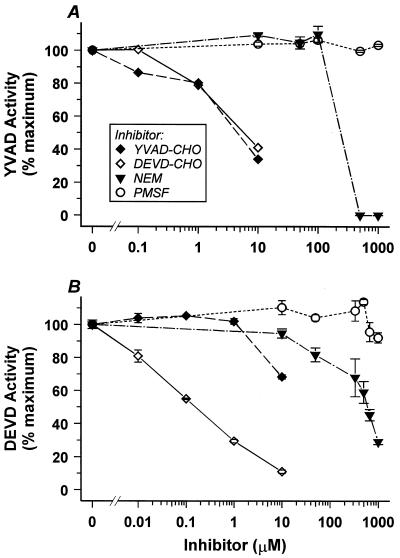
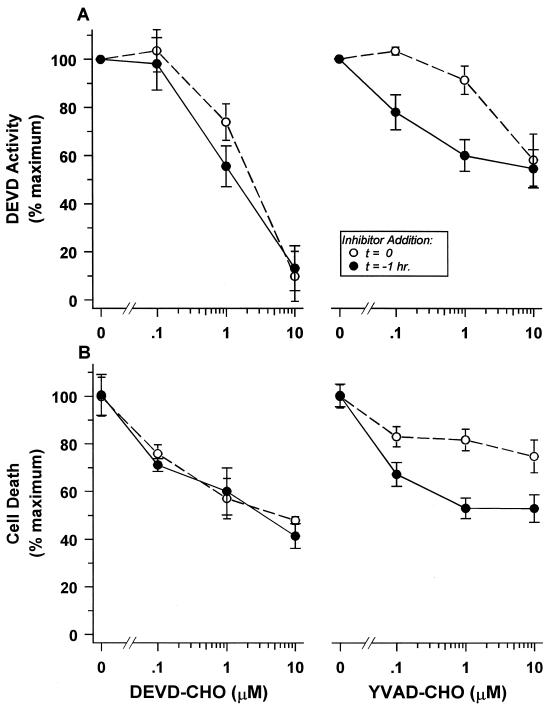
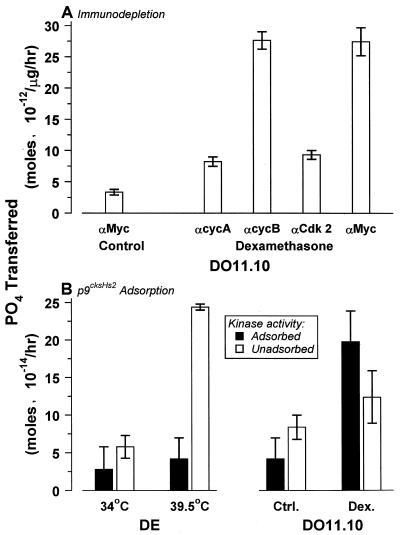
Physiological cell deaths occur ubiquitously throughout biology and have common attributes, including apoptotic morphology with mitosis-like chromatin condensation and prelytic genome digestion. The fundamental question is whether a common mechanism of dying underlies these common hallmarks of death. Here we describe evidence of such a conserved mechanism in different cells induced by distinct stimuli to undergo physiological cell death. Our genetic and quantitative biochemical analyses of T- and B-cell deaths reveal a conserved pattern of requisite components. We have dissected the role of cysteine proteases (caspases) in cell death to reflect two obligate classes of cytoplasmic activities functioning in an amplifying cascade, with upstream interleukin-1beta-converting enzyme-like proteases activating downstream caspase 3-like caspases. Bcl-2 spares cells from death by punctuating this cascade, preventing the activation of downstream caspases while leaving upstream activity undisturbed. This observation permits an operational definition of the stages of the cell death process. Upstream steps, which are necessary but not themselves lethal, are modulators of the death process. Downstream steps are effectors of, and not dissociable from, actual death; the irreversible commitment to cell death reflects the initiation of this downstream phase. In addition to caspase 3-like proteases, the effector phase of death involves the activation in the nucleus of cell cycle kinases of the cyclin-dependent kinase (Cdk) family. Nuclear recruitment and activation of Cdk components is dependent on the caspase cascade, suggesting that catastrophic Cdk activity may be the actual effector of cell death. The conservation of the cell death mechanism is not reflected in the molecular identity of its individual components, however. For example, we have detected different cyclin-Cdk pairs in different instances of cell death. The ordered course of events that we have observed in distinct cases reflects essential thematic elements of a conserved sequence of modulatory and effector activities comprising a common pathway of physiological cell death.
Figures


Similar articles
-
Defects in the ubiquitin pathway induce caspase-independent apoptosis blocked by Bcl-2.J Biol Chem. 1998 Mar 13;273(11):6121-31. doi: 10.1074/jbc.273.11.6121. J Biol Chem. 1998. PMID: 9497330
-
Conversion of Bcl-2 to a Bax-like death effector by caspases.Science. 1997 Dec 12;278(5345):1966-8. doi: 10.1126/science.278.5345.1966. Science. 1997. PMID: 9395403
-
Cellular, biochemical, and genetic analysis of mechanism of small molecule IAP inhibitors.J Biol Chem. 2004 Nov 12;279(46):48168-76. doi: 10.1074/jbc.M405022200. Epub 2004 Aug 27. J Biol Chem. 2004. PMID: 15337764
-
Proteases in apoptosis.Experientia. 1996 Oct 31;52(10-11):968-78. doi: 10.1007/BF01920106. Experientia. 1996. PMID: 8917728 Review.
-
Caspases: the proteases of the apoptotic pathway.Oncogene. 1998 Dec 24;17(25):3237-45. doi: 10.1038/sj.onc.1202581. Oncogene. 1998. PMID: 9916986 Review.
Cited by
-
Cdk2 phosphorylation of Bcl-xL after stress converts it to a pro-apoptotic protein mimicking Bax/Bak.Cell Death Discov. 2016;2:15066-. doi: 10.1038/cddiscovery.2015.66. Epub 2016 Jan 18. Cell Death Discov. 2016. PMID: 27226901 Free PMC article.
-
Cell cycle and apoptosis.Neoplasia. 2000 Jul-Aug;2(4):291-9. doi: 10.1038/sj.neo.7900101. Neoplasia. 2000. PMID: 11005563 Free PMC article. Review.
-
Caspase-dependent Cdk activity is a requisite effector of apoptotic death events.J Cell Biol. 2000 Jan 10;148(1):59-72. doi: 10.1083/jcb.148.1.59. J Cell Biol. 2000. PMID: 10629218 Free PMC article.
-
Modulation of apoptosis by the cyclin-dependent kinase inhibitor p27(Kip1).J Clin Invest. 1999 Mar;103(5):597-604. doi: 10.1172/JCI5461. J Clin Invest. 1999. PMID: 10074476 Free PMC article.
-
Caspase-2 (Nedd-2) processing and death of trophic factor-deprived PC12 cells and sympathetic neurons occur independently of caspase-3 (CPP32)-like activity.J Neurosci. 1998 Nov 15;18(22):9204-15. doi: 10.1523/JNEUROSCI.18-22-09204.1998. J Neurosci. 1998. PMID: 9801360 Free PMC article.
References
-
- Alnemri E S, Livingston D J, Nicholson D W, Salvesen G, Thornberry N A, Wong W W, Yuan J. Human ICE/Ced-3 protease nomenclature. Cell. 1996;87:171. - PubMed
-
- Baxter G D, Smith P J, Lavin M F. Molecular changes associated with induction of cell death in a human T-cell leukemia line: putative nucleases identified as histones. Biochem Biophys Res Commun. 1989;162:30–37. - PubMed
-
- Beyaert R, Kidd V J, Cornelis S, Van de Craen M, Denecker G, Lahti J M, Gururajan R, Vandenabeele P, Fiers W. Cleavage of PITSLRE kinases by ICE/CASP-1 and CPP32/CASP-3 during apoptosis induced by tumor necrosis factor. J Biol Chem. 1997;272:11694–11697. - PubMed
-
- Blomquist, J. F. Unpublished data.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials