

Helicobacter pylori disrupts epithelial barrier function in a process inhibited by protein kinase C activators
- PMID: 9596771
- PMCID: PMC108293
- DOI: 10.1128/IAI.66.6.2943-2950.1998
Helicobacter pylori disrupts epithelial barrier function in a process inhibited by protein kinase C activators
Abstract
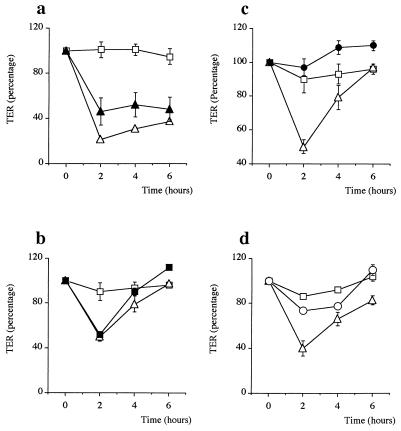
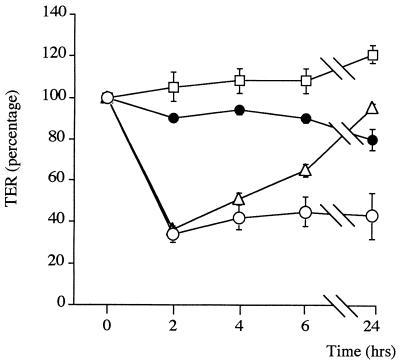
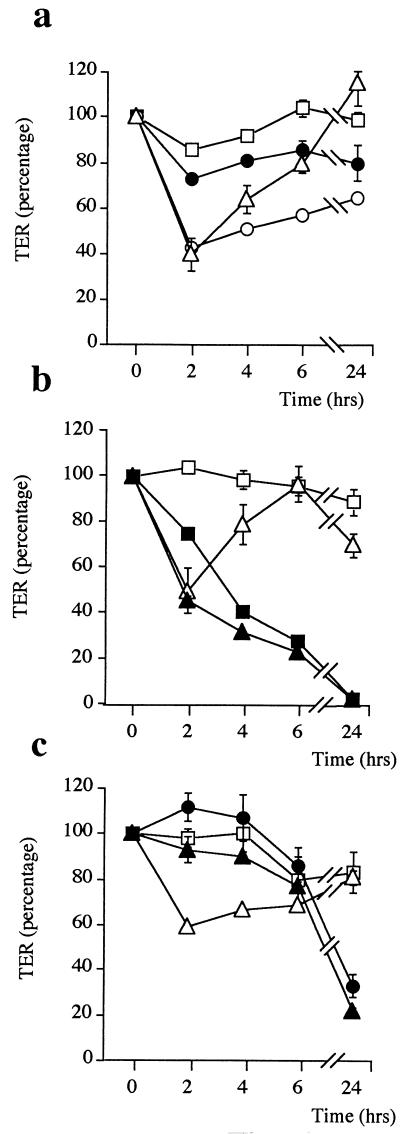
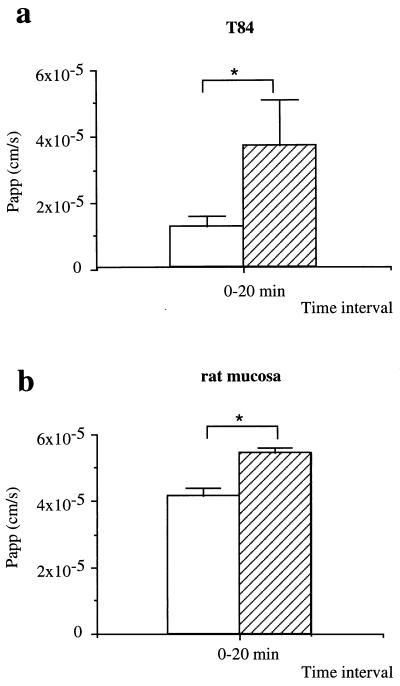
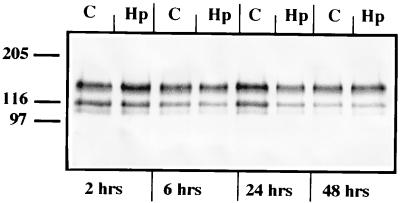
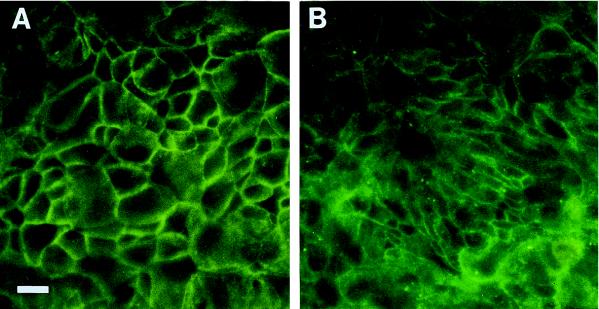
Helicobacter pylori colonizes the gastric mucosa, and the infection is related to the development of diverse gastric pathologies, possibly by directly or indirectly affecting epithelial-cell function. We analyzed the influence of the bacteria on transepithelial electrical resistance (TER) on a model tight epithelium, T84, grown to confluence in permeable filters. H. pylori sonicates produced a dramatic decrease in TER after 1 to 2 h of exposure, while sonicates from other bacteria did not induce a significant reduction of TER. The effect induced by sonicates was mimicked by a water-soluble fraction from the bacterial surface, was not reproducible with isolated lipopolysaccharide, and was concomitant with a significant increase in the paracellular permeability of the marker molecule [14C]mannitol. Furthermore, H. pylori sonicates also provoked a significant increase in permeability to [14C]mannitol across rat gastric mucosa in vitro. The sonicate-induced decrease in TER in T84 monolayers was inhibited by the protein kinase C (PKC) activator phorbol myristate acetate. As PKC is directly involved in tight junction regulation, we suggest that H. pylori may induce intracellular signalling events counteracting PKC effects. Following long-term H. pylori stimulation, epithelial monolayers regained baseline resistance values slowly after 24 h. The resistance recovery process was inhibited by cycloheximide, indicating its dependency upon protein synthesis. No association between resistance variation and E-cadherin protein levels was observed. These results indicate that H. pylori alters in vitro the barrier properties of the epithelium, probably by generating cell signalling events counteracting the normal function of PKC. This increased permeability may provide a potential mechanism by which H. pylori antigens can reach the gastric lamina propria, thereby activating the mucosal immune system.
Figures




References
-
- Aghib D, McCrea P. The E-cadherin complex contains the src substrate p120. Exp Cell Res. 1995;218:359–369. - PubMed
-
- Anderson J, Itallie C V. Tight junctions and the molecular basis for regulation of paracellular permeability. Am J Physiol. 1995;269:G467–G475. - PubMed
-
- Balda M, Gonzales-Mariscal L, Contreras R, Macias-Silva M, Torres-Markez M, Garcia-Sainz J, Cereijido M. Assembly and sealing of tight junctions: possible participation of G-proteins, phospholipase C, protein kinase C and calmodulin. J Membr Biol. 1991;122:193–202. - PubMed
-
- Borenfreund E, Puerner J. A simple quantitative procedure using monolayer cultures for cytotoxicity assays (9HTD/NR-90) J Tissue Cult Methods. 1984;9:7–9.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
