

Measuring genome evolution
- PMID: 9600883
- PMCID: PMC34486
- DOI: 10.1073/pnas.95.11.5849
Measuring genome evolution
Abstract
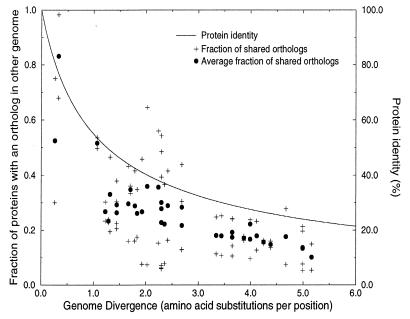
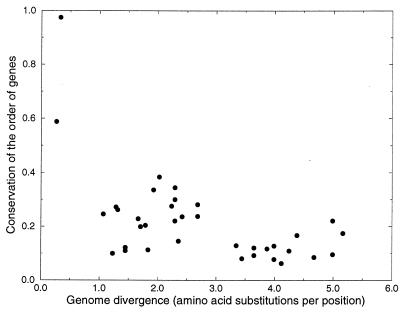
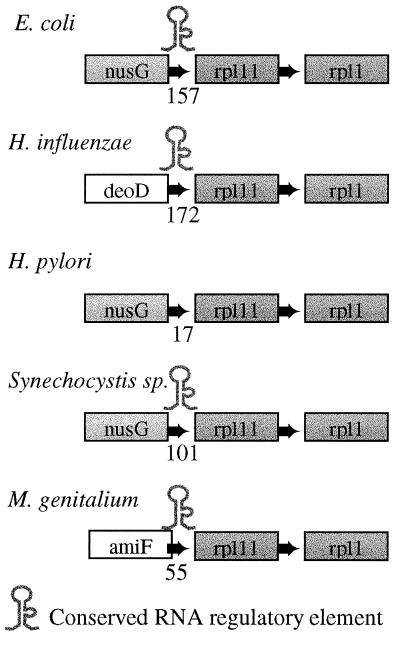
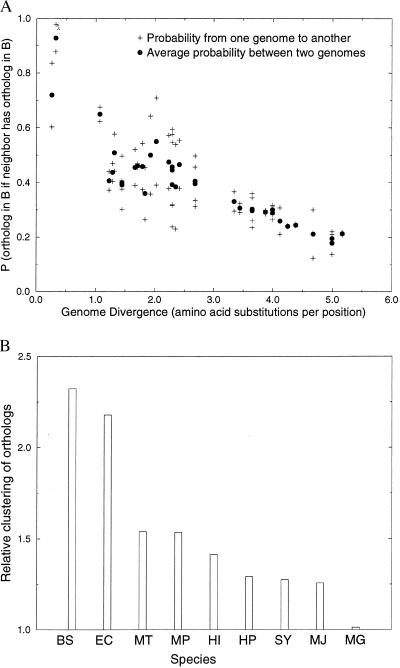
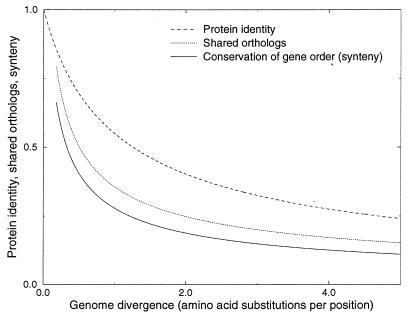
The determination of complete genome sequences provides us with an opportunity to describe and analyze evolution at the comprehensive level of genomes. Here we compare nine genomes with respect to their protein coding genes at two levels: (i) we compare genomes as "bags of genes" and measure the fraction of orthologs shared between genomes and (ii) we quantify correlations between genes with respect to their relative positions in genomes. Distances between the genomes are related to their divergence times, measured as the number of amino acid substitutions per site in a set of 34 orthologous genes that are shared among all the genomes compared. We establish a hierarchy of rates at which genomes have changed during evolution. Protein sequence identity is the most conserved, followed by the complement of genes within the genome. Next is the degree of conservation of the order of genes, whereas gene regulation appears to evolve at the highest rate. Finally, we show that some genomes are more highly organized than others: they show a higher degree of the clustering of genes that have orthologs in other genomes.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
