

Electrostatic steering and ionic tethering in enzyme-ligand binding: insights from simulations
- PMID: 9600896
- PMCID: PMC34177
- DOI: 10.1073/pnas.95.11.5942
Electrostatic steering and ionic tethering in enzyme-ligand binding: insights from simulations
Abstract
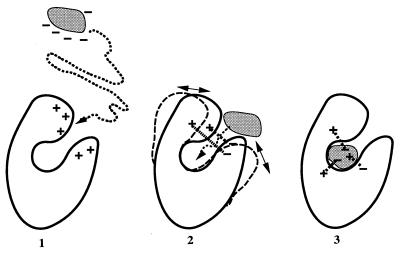
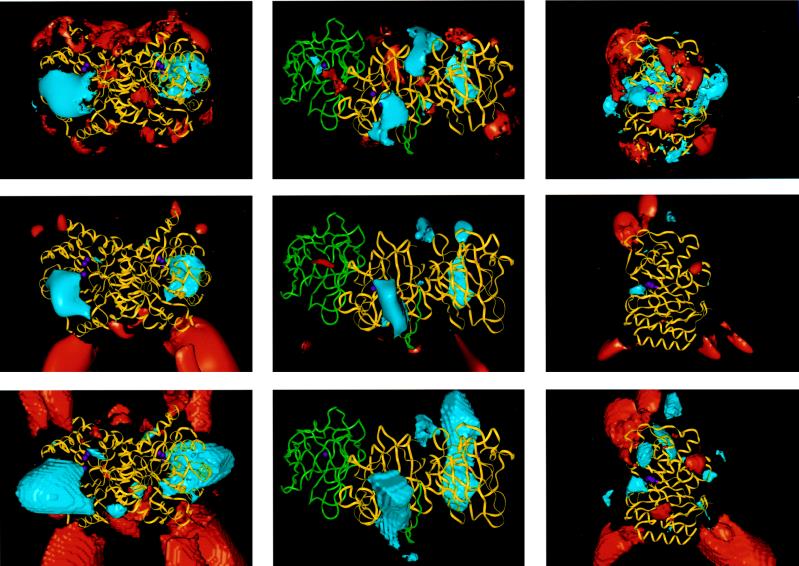
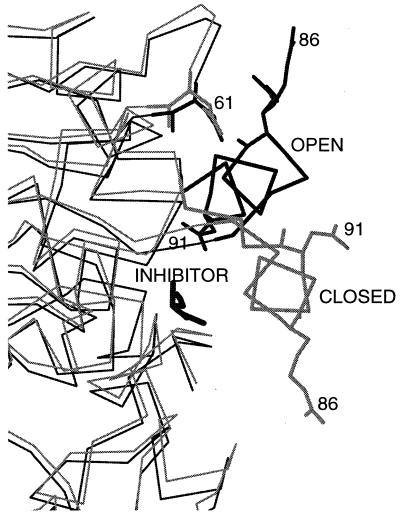
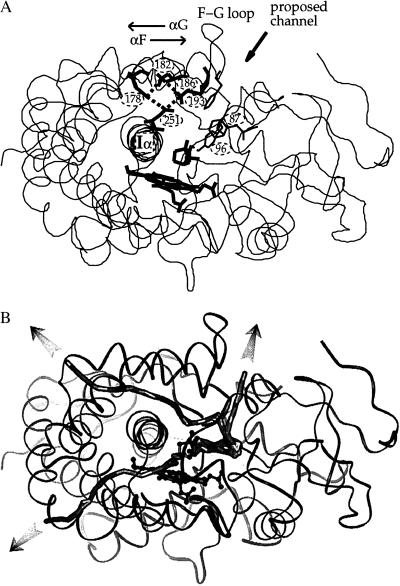
To bind at an enzyme's active site, a ligand must diffuse or be transported to the enzyme's surface, and, if the binding site is buried, the ligand must diffuse through the protein to reach it. Although the driving force for ligand binding is often ascribed to the hydrophobic effect, electrostatic interactions also influence the binding process of both charged and nonpolar ligands. First, electrostatic steering of charged substrates into enzyme active sites is discussed. This is of particular relevance for diffusion-influenced enzymes. By comparing the results of Brownian dynamics simulations and electrostatic potential similarity analysis for triose-phosphate isomerases, superoxide dismutases, and beta-lactamases from different species, we identify the conserved features responsible for the electrostatic substrate-steering fields. The conserved potentials are localized at the active sites and are the primary determinants of the bimolecular association rates. Then we focus on a more subtle effect, which we will refer to as "ionic tethering." We explore, by means of molecular and Brownian dynamics simulations and electrostatic continuum calculations, how salt links can act as tethers between structural elements of an enzyme that undergo conformational change upon substrate binding, and thereby regulate or modulate substrate binding. This is illustrated for the lipase and cytochrome P450 enzymes. Ionic tethering can provide a control mechanism for substrate binding that is sensitive to the electrostatic properties of the enzyme's surroundings even when the substrate is nonpolar.
Figures
References
-
- Davis M E, Madura J D, Sines J, Luty B A, Allison S A, McCammon J A. Methods Enzymol. 1991;202:473–497. - PubMed
-
- Tan R C, Truong T N, McCammon J A, Sussman J L. Biochemistry. 1993;32:401–403. - PubMed
-
- Wade R C. Biochem Soc Trans. 1996;24:254–259. - PubMed
-
- Madura J D, Briggs J M, Wade R C, Gabdoulline R R. In: Encyclopedia of Computational Chemistry. von Rague Schleyer P, Allinger N L, Clark T, Gasteiger J, Kollman P A, Schaefer H F, editors. Chichester, U.K.: Wiley; 1998. , in press.
-
- Getzoff E D, Cabelli D E, Fisher C L, Parge H E, Viezzoli M S, Banci L, Hallewell R A. Nature (London) 1992;358:347–351. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
