

Mutations in the chloride-bicarbonate exchanger gene AE1 cause autosomal dominant but not autosomal recessive distal renal tubular acidosis
- PMID: 9600966
- PMCID: PMC27686
- DOI: 10.1073/pnas.95.11.6337
Mutations in the chloride-bicarbonate exchanger gene AE1 cause autosomal dominant but not autosomal recessive distal renal tubular acidosis
Abstract
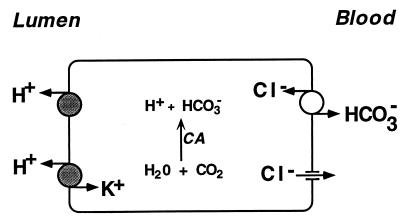
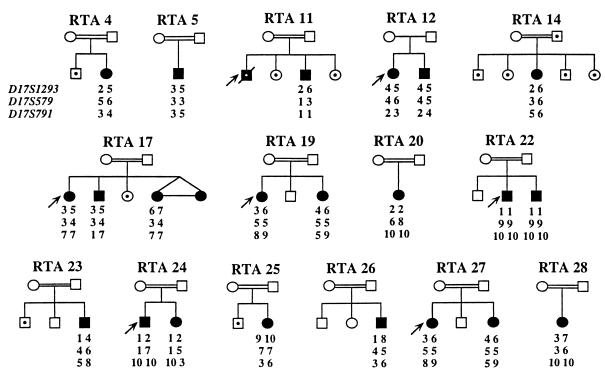
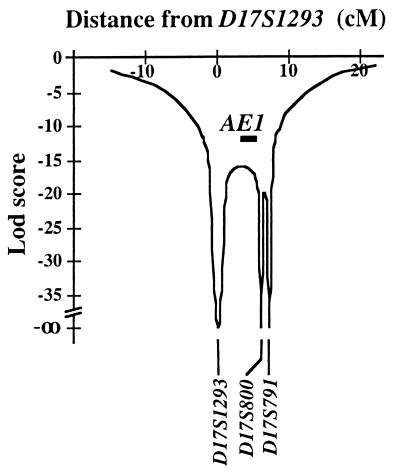
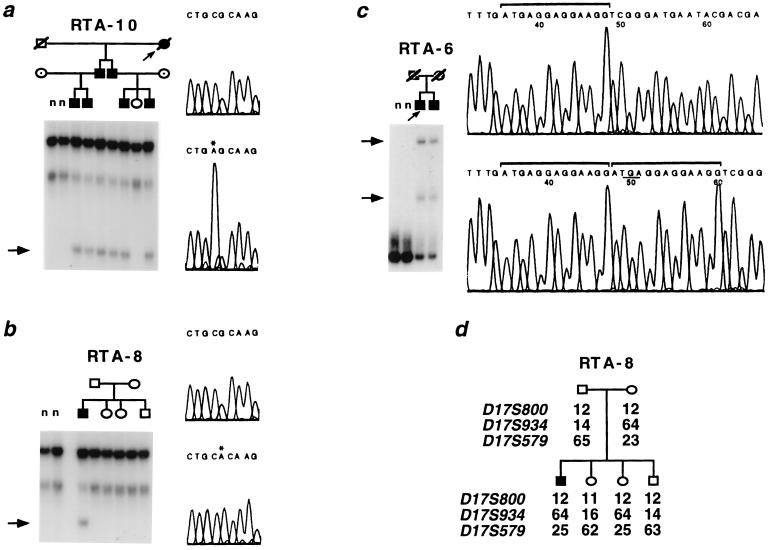
Primary distal renal tubular acidosis (dRTA) is characterized by reduced ability to acidify urine, variable hyperchloremic hypokalemic metabolic acidosis, nephrocalcinosis, and nephrolithiasis. Kindreds showing either autosomal dominant or recessive transmission are described. Mutations in the chloride-bicarbonate exchanger AE1 have recently been reported in four autosomal dominant dRTA kindreds, three of these altering codon Arg589. We have screened 26 kindreds with primary dRTA for mutations in AE1. Inheritance was autosomal recessive in seventeen kindreds, autosomal dominant in one, and uncertain due to unknown parental phenotype or sporadic disease in eight kindreds. No mutations in AE1 were detected in any of the autosomal recessive kindreds, and analysis of linkage showed no evidence of linkage of recessive dRTA to AE1. In contrast, heterozygous mutations in AE1 were identified in the one known dominant dRTA kindred, in one sporadic case, and one kindred with two affected brothers. In the dominant kindred, the mutation Arg-589/Ser cosegregated with dRTA in the extended pedigree. An Arg-589/His mutation in the sporadic case proved to be a de novo mutation. In the third kindred, affected brothers both have an intragenic 13-bp duplication resulting in deletion of the last 11 amino acids of AE1. These mutations were not detected in 80 alleles from unrelated normal individuals. These findings underscore the key role of Arg-589 and the C terminus in normal AE1 function, and indicate that while mutations in AE1 cause autosomal dominant dRTA, defects in this gene are not responsible for recessive disease.
Figures
Similar articles
-
Autosomal recessive distal renal tubular acidosis caused by G701D mutation of anion exchanger 1 gene.Am J Kidney Dis. 2002 Jul;40(1):21-9. doi: 10.1053/ajkd.2002.33909. Am J Kidney Dis. 2002. PMID: 12087557
-
Hereditary distal renal tubular acidosis: new understandings.Annu Rev Med. 2001;52:471-84. doi: 10.1146/annurev.med.52.1.471. Annu Rev Med. 2001. PMID: 11160790 Review.
-
Autosomal dominant distal renal tubular acidosis is associated in three families with heterozygosity for the R589H mutation in the AE1 (band 3) Cl-/HCO3- exchanger.J Biol Chem. 1998 Mar 13;273(11):6380-8. doi: 10.1074/jbc.273.11.6380. J Biol Chem. 1998. PMID: 9497368
-
Atypical distal renal tubular acidosis confirmed by mutation analysis.Pediatr Nephrol. 2000 Dec;15(3-4):201-4. doi: 10.1007/s004670000454. Pediatr Nephrol. 2000. PMID: 11149111
-
[Primary distal renal tubular acidosis].Ann Biol Clin (Paris). 2009 Mar-Apr;67(2):135-40. doi: 10.1684/abc.2009.0307. Ann Biol Clin (Paris). 2009. PMID: 19297287 Review. French.
Cited by
-
Collecting duct intercalated cell function and regulation.Clin J Am Soc Nephrol. 2015 Feb 6;10(2):305-24. doi: 10.2215/CJN.08880914. Epub 2015 Jan 28. Clin J Am Soc Nephrol. 2015. PMID: 25632105 Free PMC article. Review.
-
A single nucleotide polymorphism in kidney anion exchanger 1 gene is associated with incomplete type 1 renal tubular acidosis.Sci Rep. 2016 Oct 21;6:35841. doi: 10.1038/srep35841. Sci Rep. 2016. PMID: 27767102 Free PMC article.
-
Evidence from renal proximal tubules that HCO3- and solute reabsorption are acutely regulated not by pH but by basolateral HCO3- and CO2.Proc Natl Acad Sci U S A. 2005 Mar 8;102(10):3875-80. doi: 10.1073/pnas.0500423102. Epub 2005 Feb 22. Proc Natl Acad Sci U S A. 2005. PMID: 15728388 Free PMC article.
-
Autosomal dominant distal renal tubular acidosis caused by a mutation in the anion exchanger 1 gene in a Japanese family.CEN Case Rep. 2015 Nov;4(2):218-222. doi: 10.1007/s13730-015-0172-3. Epub 2015 Mar 4. CEN Case Rep. 2015. PMID: 28509104 Free PMC article.
-
Adaptor protein 1 complexes regulate intracellular trafficking of the kidney anion exchanger 1 in epithelial cells.Am J Physiol Cell Physiol. 2012 Sep 1;303(5):C554-66. doi: 10.1152/ajpcell.00124.2012. Epub 2012 Jun 27. Am J Physiol Cell Physiol. 2012. PMID: 22744004 Free PMC article.
References
-
- Rodriguez-Soriano J, Vallo A. Pediatr Nephrol. 1990;4:268–275. - PubMed
-
- Batlle D, Flores G. Am J Kidney Dis. 1996;6:896–915. - PubMed
-
- Brown M T, Cunningham M J, Ingelfinger J R, Becker A N. Arch Otolaryngol Head Neck Surg. 1993;119:458–460. - PubMed
-
- Stone D K, Xie X S. Kidney Int. 1988;33:767–774. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
