

A physiological role of the adenosine A3 receptor: sustained cardioprotection
- PMID: 9618527
- PMCID: PMC22715
- DOI: 10.1073/pnas.95.12.6995
A physiological role of the adenosine A3 receptor: sustained cardioprotection
Abstract
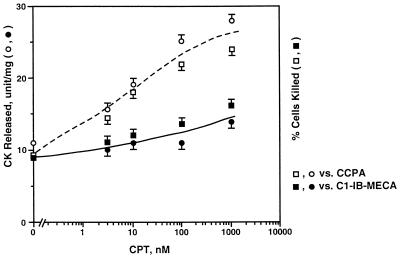
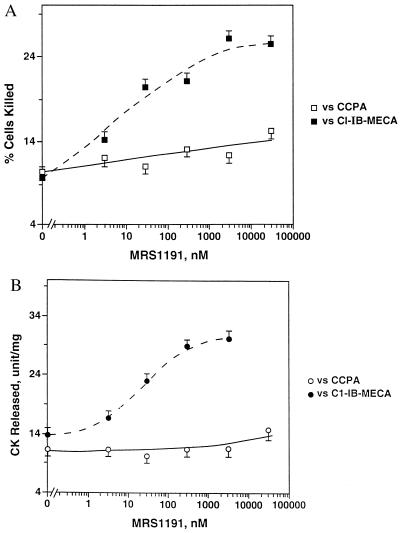
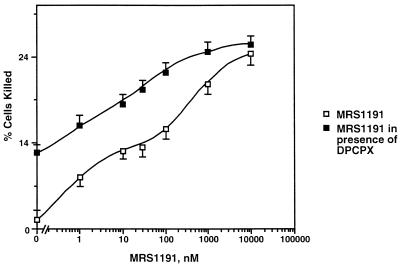
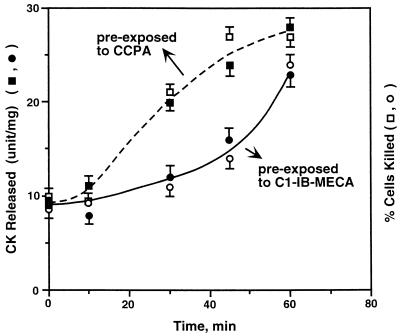
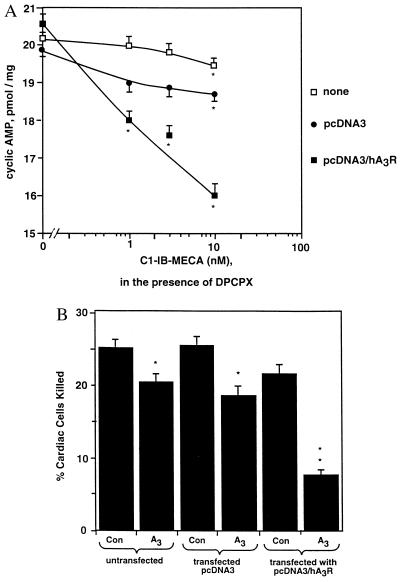
Adenosine released during cardiac ischemia exerts a potent, protective effect in the heart. A newly recognized adenosine receptor, the A3 subtype, is expressed on the cardiac ventricular cell, and its activation protects the ventricular heart cell against injury during a subsequent exposure to ischemia. A cultured chicken ventricular myocyte model was used to investigate the cardioprotective role of a novel adenosine A3 receptor. The protection mediated by prior activation of A3 receptors exhibits a significantly longer duration than that produced by activation of the adenosine A1 receptor. Prior exposure of the myocytes to brief ischemia also protected them against injury sustained during a subsequent exposure to prolonged ischemia. The adenosine A3 receptor-selective antagonist 3-ethyl 5-benzyl-2-methyl-6-phenyl-4-phenylethynyl-1, 4-(+/-)-dihydropyridine-3,5-dicarboxylate (MRS1191) caused a biphasic inhibition of the protective effect of the brief ischemia. The concomitant presence of the A1 receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) converted the MRS1191-induced dose inhibition curve to a monophasic one. The combined presence of both antagonists abolished the protective effect induced by the brief ischemia. Thus, activation of both A1 and A3 receptors is required to mediate the cardioprotective effect of the brief ischemia. Cardiac atrial cells lack native A3 receptors and exhibit a shorter duration of cardioprotection than do ventricular cells. Transfection of atrial cells with cDNA encoding the human adenosine A3 receptor causes a sustained A3 agonist-mediated cardioprotection. The study indicates that cardiac adenosine A3 receptor mediates a sustained cardioprotective function and represents a new cardiac therapeutic target.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
