

Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness
- PMID: 9618555
- PMCID: PMC22772
- DOI: 10.1073/pnas.95.12.7157
Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness
Abstract
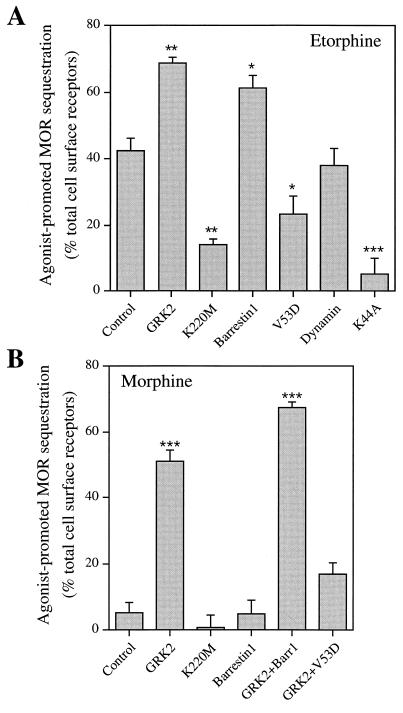
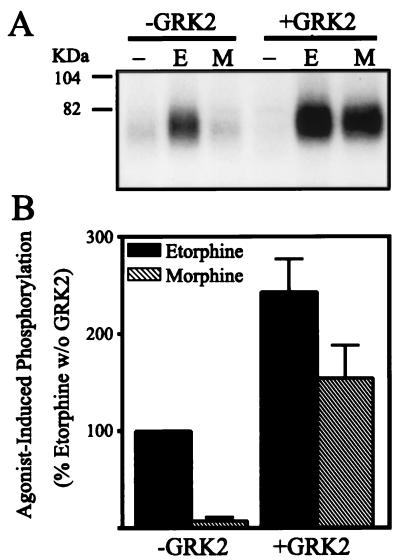
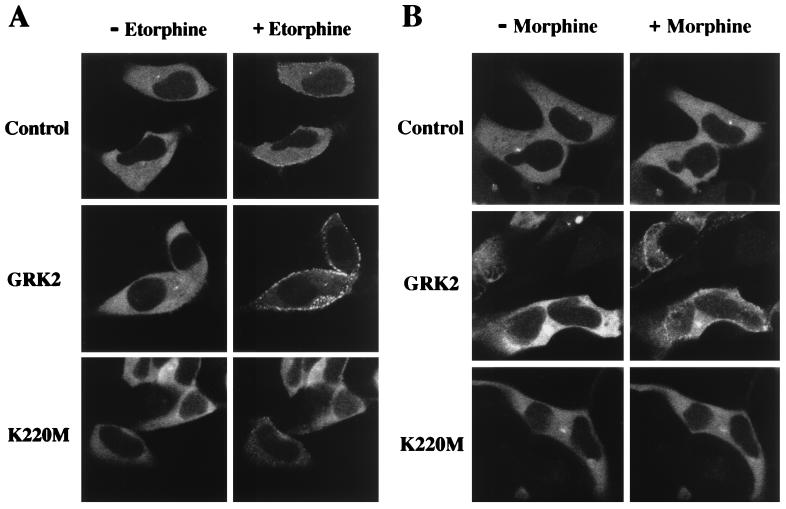
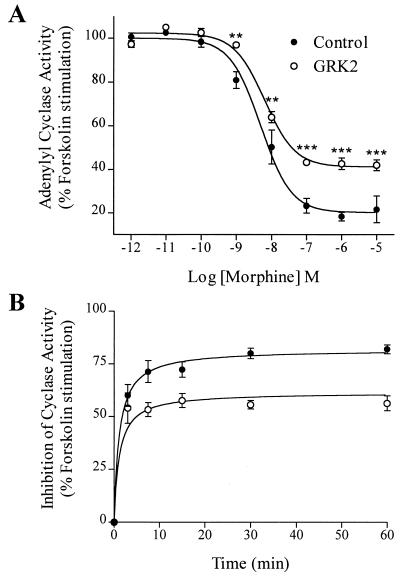
The G protein-coupled mu-opioid receptor (mu OR) mediates the physiological effects of endogenous opioid peptides as well as the structurally distinct opioid alkaloids morphine and etorphine. An intriguing feature of mu OR signaling is the differential receptor trafficking and desensitization properties following activation by distinct agonists, which have been proposed as possible mechanisms related to opioid tolerance. Here we report that the ability of distinct opioid agonists to differentially regulate mu OR internalization and desensitization is related to their ability to promote G protein-coupled receptor kinase (GRK)-dependent phosphorylation of the mu OR. Although both etorphine and morphine effectively activate the mu OR, only etorphine elicits robust mu OR phosphorylation followed by plasma membrane translocation of beta-arrestin and dynamin-dependent receptor internalization. In contrast, corresponding to its inability to cause mu OR internalization, morphine is unable to either elicit mu OR phosphorylation or stimulate beta-arrestin translocation. However, upon the overexpression of GRK2, morphine gains the capacity to induce mu OR phosphorylation, accompanied by the rescue of beta-arrestin translocation and receptor sequestration. Moreover, overexpression of GRK2 also leads to an attenuation of morphine-mediated inhibition of adenylyl cyclase. These findings point to the existence of marked differences in the ability of different opioid agonists to promote mu OR phosphorylation by GRK. These differences may provide the molecular basis underlying the different analgesic properties of opioid agonists and contribute to the distinct ability of various opioids to induce drug tolerance.
Figures
References
-
- Cherny N I. Drugs. 1996;51:713–737. - PubMed
-
- Piros E T, Hales T G, Evans C J. Neurochem Res. 1996;21:1277–1285. - PubMed
-
- Standifer K M, Pasternak G W. Cell Signalling. 1997;9:237–248. - PubMed
-
- Raynor K, Kong H, Law S, Heerding J, Tallent M, Livingston F, Hines J, Reisine T. NIDA Res Monogr. 1996;161:83–103. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
