

Appropriate expression of filamentous phage f1 DNA replication genes II and X requires RNase E-dependent processing and separate mRNAs
- PMID: 9620980
- PMCID: PMC107831
- DOI: 10.1128/JB.180.12.3245-3249.1998
Appropriate expression of filamentous phage f1 DNA replication genes II and X requires RNase E-dependent processing and separate mRNAs
Abstract
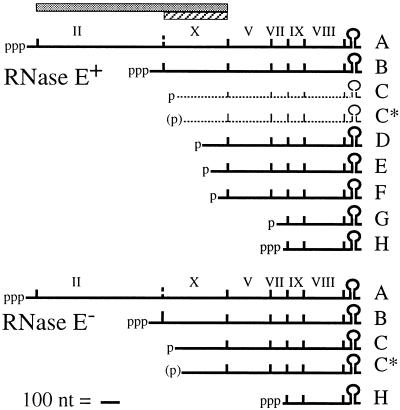
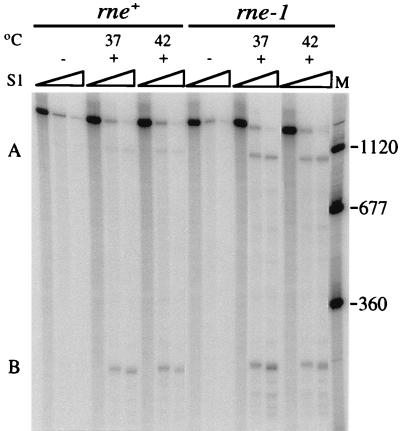
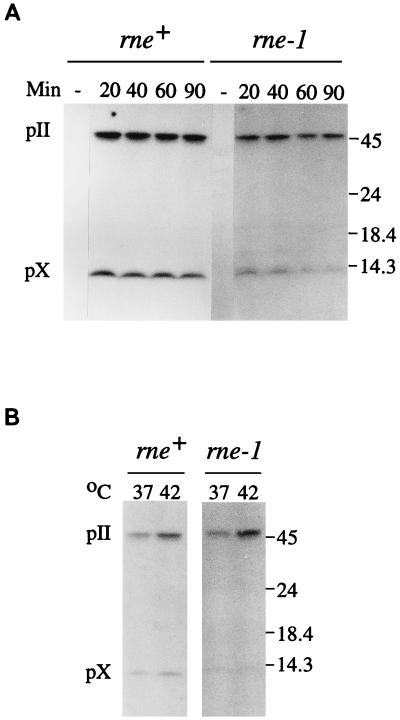
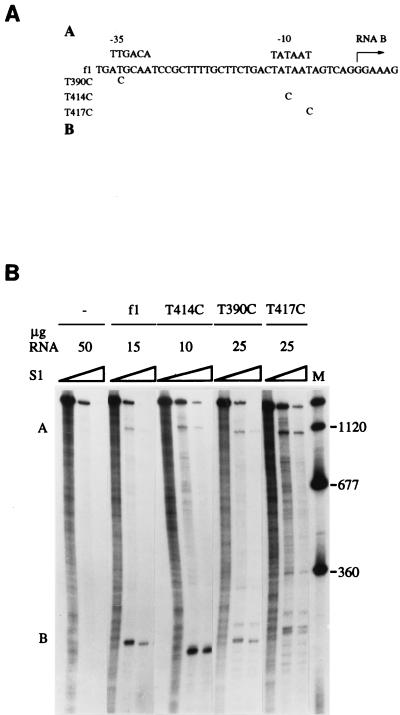
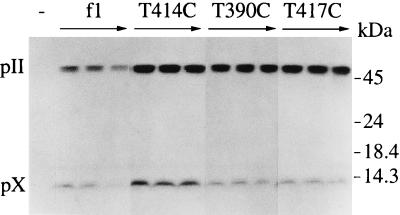
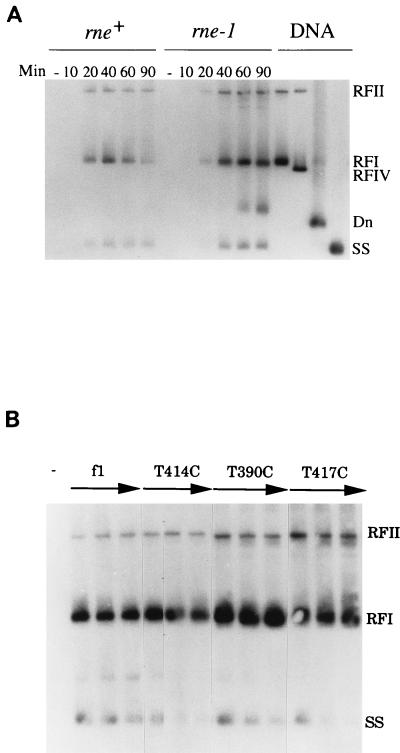
The products of in-frame overlapping genes II and X carried by the filamentous phage f1 genome are proteins with required but opposing functions in phage DNA replication. Their normal relative levels are important for continuous production of phage DNA without killing infected Escherichia coli hosts. Here we identify several factors responsible for determining the relative levels of pII and pX and that, if perturbed, alter the normal distribution of the phage DNA species in infected hosts. Translation of the two proteins is essentially relegated to separate mRNAs. The mRNAs encoding genes II and X are also differentially sensitive to cleavage dependent on rne, the gene encoding the only E. coli endo-RNase known to have a global role in mRNA stability. Whereas pII levels are limited at the level of mRNA stability, normal pX levels require transcription in sufficient amounts from the promoter for the smaller mRNA encoding only pX.
Figures
Similar articles
-
Functional analysis of filamentous phage f1 mRNA processing sites.RNA. 1996 Dec;2(12):1286-94. RNA. 1996. PMID: 8972776 Free PMC article.
-
In-frame overlapping genes: the challenges for regulating gene expression.Mol Microbiol. 2007 Feb;63(4):1158-72. doi: 10.1111/j.1365-2958.2006.05572.x. Mol Microbiol. 2007. PMID: 17238928
-
Roles of polyadenylation and nucleolytic cleavage in the filamentous phage mRNA processing and decay pathways in Escherichia coli.RNA. 1999 Jul;5(7):972-85. doi: 10.1017/s1355838299990398. RNA. 1999. PMID: 10411140 Free PMC article.
-
The multifaceted roles of the RNA processing enzyme ribonuclease III.Indian J Biochem Biophys. 1996 Aug;33(4):253-60. Indian J Biochem Biophys. 1996. PMID: 8936814 Review.
-
Posttranscriptional control of the lysogenic pathway in bacteriophage lambda.Prog Nucleic Acid Res Mol Biol. 1993;46:37-49. doi: 10.1016/s0079-6603(08)61017-x. Prog Nucleic Acid Res Mol Biol. 1993. PMID: 8234786 Review. No abstract available.
Cited by
-
The ribosome binding site of a mini-ORF protects a T3SS mRNA from degradation by RNase E.Mol Microbiol. 2012 Dec;86(5):1167-82. doi: 10.1111/mmi.12050. Epub 2012 Oct 12. Mol Microbiol. 2012. PMID: 23043360 Free PMC article.
-
Transcriptional analysis of the sfa determinant revealing mmRNA processing events in the biogenesis of S fimbriae in pathogenic Escherichia coli.J Bacteriol. 2003 Jan;185(2):620-9. doi: 10.1128/JB.185.2.620-629.2003. J Bacteriol. 2003. PMID: 12511509 Free PMC article.
-
Global analysis of Escherichia coli RNA degradosome function using DNA microarrays.Proc Natl Acad Sci U S A. 2004 Mar 2;101(9):2758-63. doi: 10.1073/pnas.0308747101. Epub 2004 Feb 23. Proc Natl Acad Sci U S A. 2004. PMID: 14981237 Free PMC article.
-
Filamentous phages: masters of a microbial sharing economy.EMBO Rep. 2019 Jun;20(6):e47427. doi: 10.15252/embr.201847427. Epub 2019 Apr 5. EMBO Rep. 2019. PMID: 30952693 Free PMC article. Review.
-
Post-transcriptional processing of the LEE4 operon in enterohaemorrhagic Escherichia coli.Mol Microbiol. 2009 Jan;71(2):273-90. doi: 10.1111/j.1365-2958.2008.06530.x. Epub 2008 Nov 4. Mol Microbiol. 2009. PMID: 19019141 Free PMC article.
References
-
- Cashman J S, Webster R E, Steege D A. Transcription of bacteriophage f1: the major in vivo RNAs. J Biol Chem. 1980;255:2554–2562. - PubMed
-
- Cohen S N, McDowall K J. RNase E: still a wonderfully mysterious enzyme. Mol Microbiol. 1997;23:1099–1106. - PubMed
-
- Fulford W, Model P. Specificity of translational regulation by two DNA binding proteins. J Mol Biol. 1984;173:211–226. - PubMed
-
- Fulford W, Model P. Regulation of bacteriophage f1 DNA replication. I. New functions for genes II and X. J Mol Biol. 1988;203:49–62. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
