

Retroviral diversity and distribution in vertebrates
- PMID: 9621058
- PMCID: PMC110400
- DOI: 10.1128/JVI.72.7.5955-5966.1998
Retroviral diversity and distribution in vertebrates
Abstract
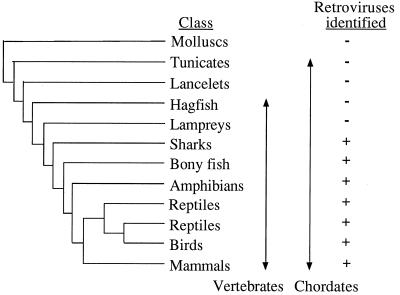
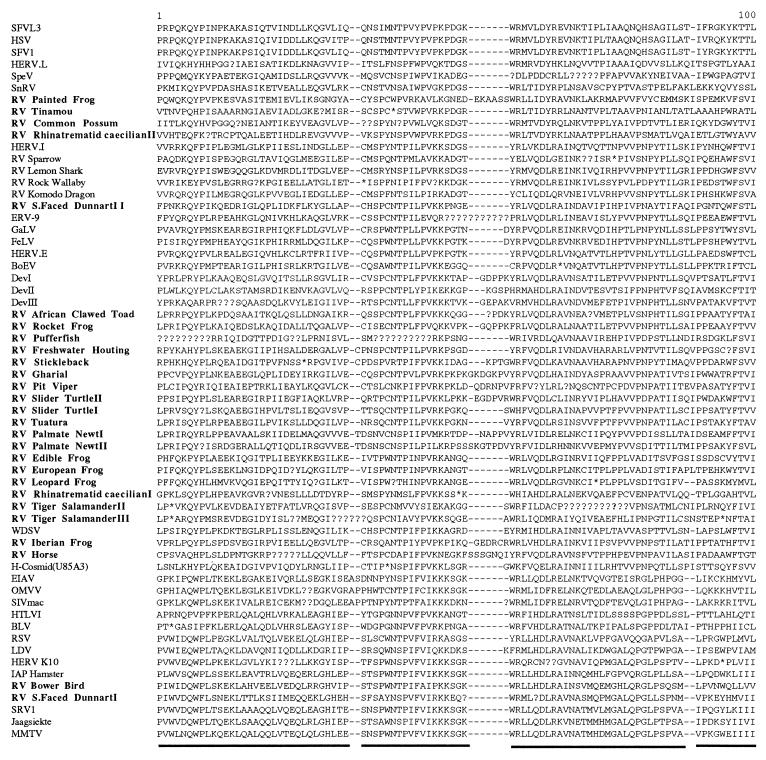
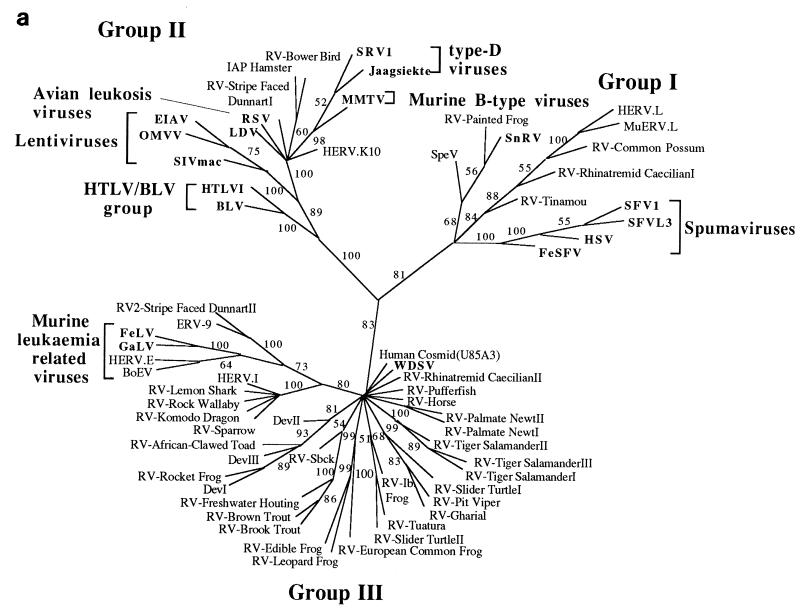
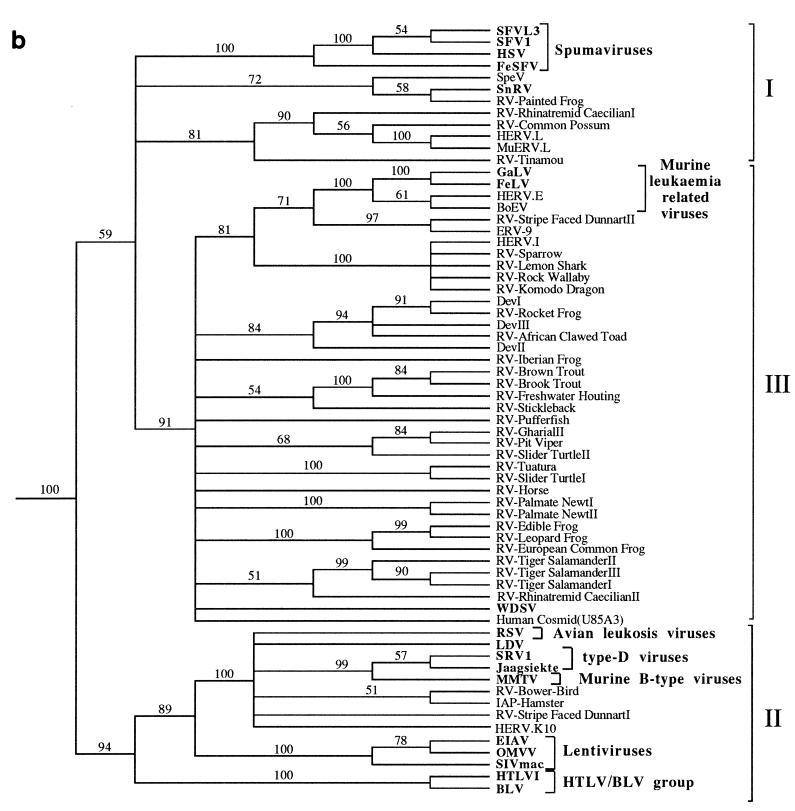
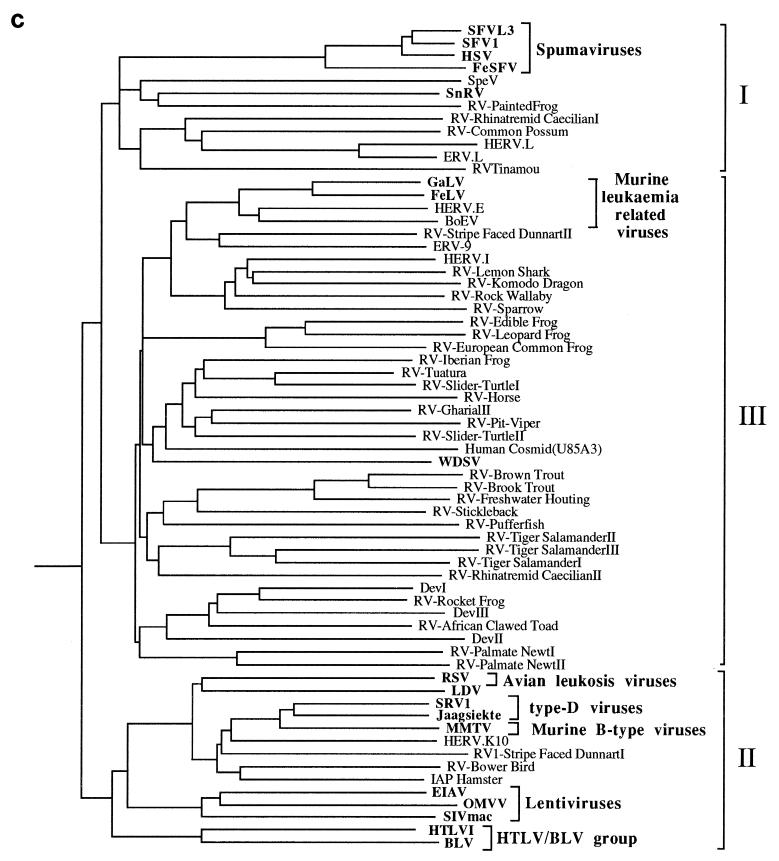
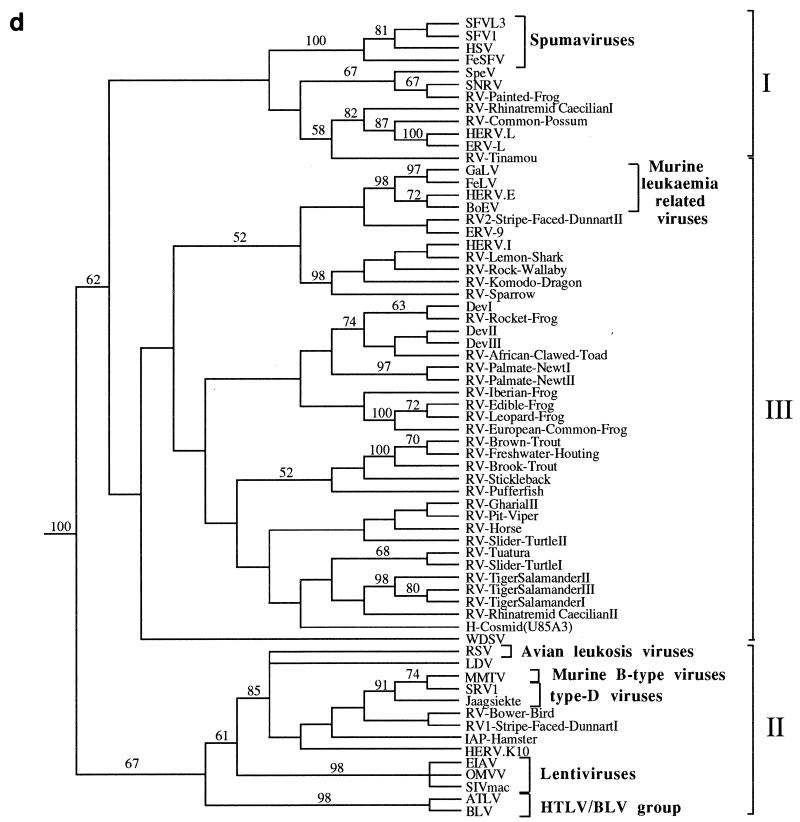
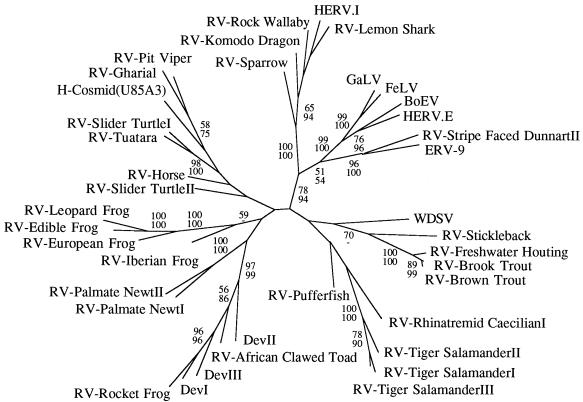
We used the PCR to screen for the presence of endogenous retroviruses within the genomes of 18 vertebrate orders across eight classes, concentrating on reptilian, amphibian, and piscine hosts. Thirty novel retroviral sequences were isolated and characterized by sequencing approximately 1 kb of their encoded protease and reverse transcriptase genes. Isolation of novel viruses from so many disparate hosts suggests that retroviruses are likely to be ubiquitous within all but the most basal vertebrate classes and, furthermore, gives a good indication of the overall retroviral diversity within vertebrates. Phylogenetic analysis demonstrated that viruses clustering with (but not necessarily closely related to) the spumaviruses and murine leukemia viruses are widespread and abundant in vertebrate genomes. In contrast, we were unable to identify any viruses from hosts outside of mammals and birds which grouped with the other five currently recognized retroviral genera: the lentiviruses, human T-cell leukemia-related viruses, avian leukemia virus-related retroviruses, type D retroviruses, and mammalian type B retroviruses. There was also some indication that viruses isolated from individual vertebrate classes tended to cluster together in phylogenetic reconstructions. This implies that the horizontal transmission of at least some retroviruses, between some vertebrate classes, occurs relatively infrequently. It is likely that many of the retroviral sequences described here are distinct enough from those of previously characterized viruses to represent novel retroviral genera.
Figures

References
-
- Altschul S F, Gish W, Miller W, Myers E W, Lipman D L. Basic alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
-
- Bénit L, de Parseval N, Casella J F, Callebaut I, Cordonnier A, Heidmann T. Cloning of a new murine endogenous retrovirus, MuERV- L, with strong similarity to the human HERV-L element and with a gag coding sequence closely related to the Fv1 restriction gene. J Virol. 1997;71:5652–5657. - PMC - PubMed
-
- Chen Y C, Cui Z, Lee L F, Witter R L. Serological differences among non-defective reticuloendotheliosis viruses. Arch Virol. 1987;93:233–245. - PubMed
-
- Coffin J M. Structure and classification of retroviruses. In: Levy J A, editor. The Retroviridae. Vol. 1. New York, N.Y: Plenum Press; 1992. pp. 19–49.
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
