

Primary activation of interferon A and interferon B gene transcription by interferon regulatory factor 3
- PMID: 9707562
- PMCID: PMC21423
- DOI: 10.1073/pnas.95.17.9837
Primary activation of interferon A and interferon B gene transcription by interferon regulatory factor 3
Abstract
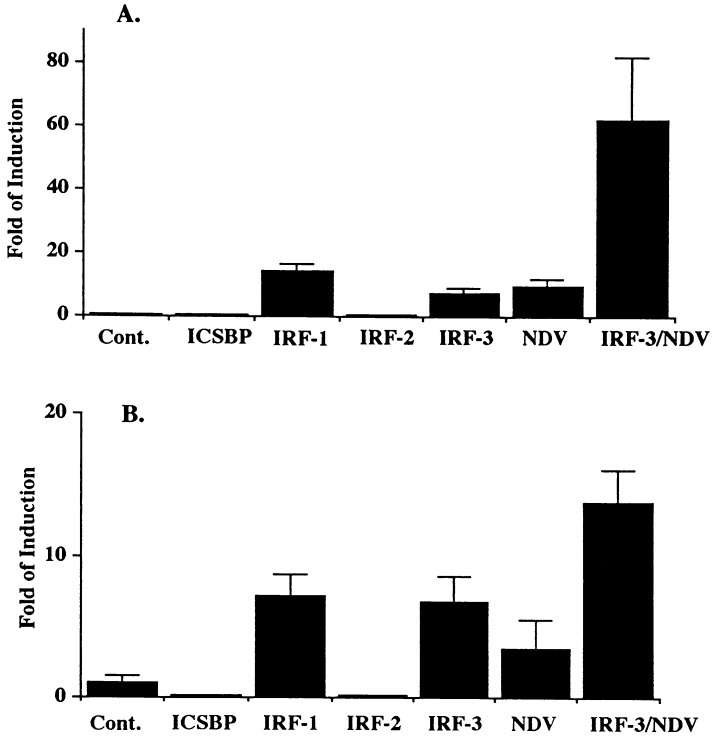
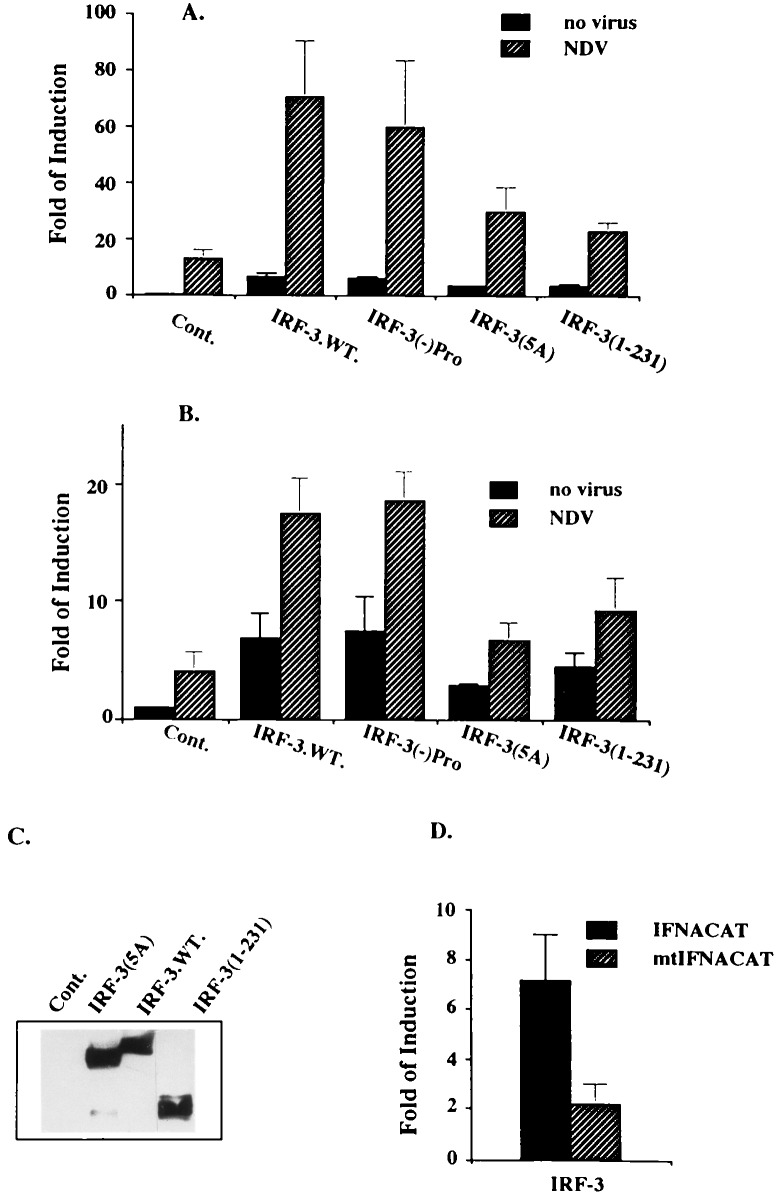
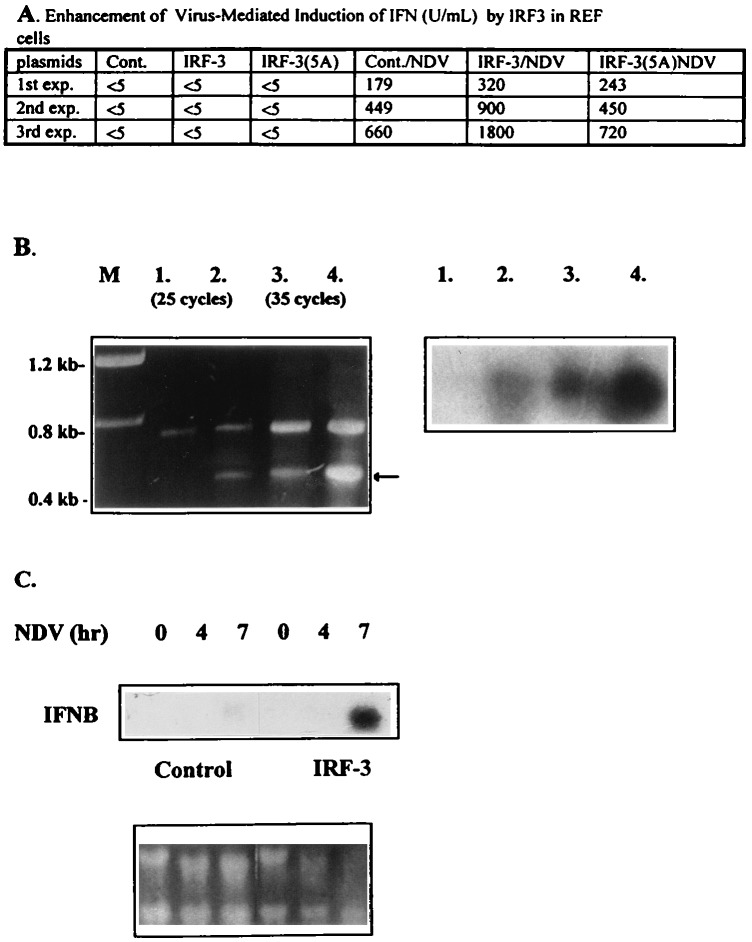
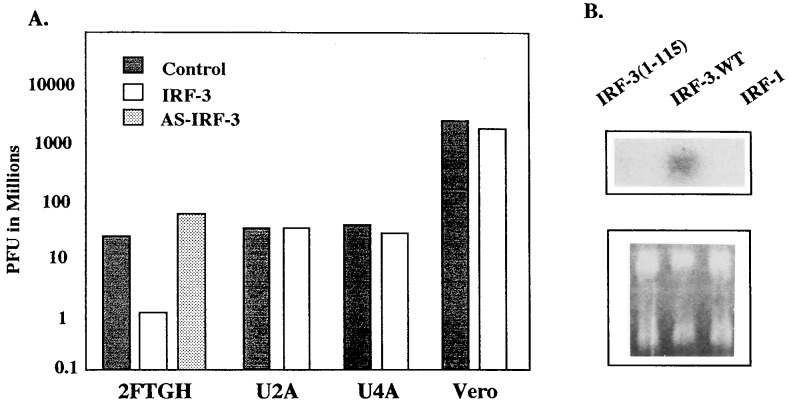
The family of interferon (IFN) regulatory factors (IRFs) encodes DNA-binding transcription factors, some of which function as modulators of virus-induced signaling. The IRF-3 gene is constitutively expressed in many tissues and cell types, and neither virus infection nor IFN treatment enhances its transcription. In infected cells, however, IRF-3 protein is phosphorylated at the carboxyl terminus, which facilitates its binding to the CBP/p300 coactivator. In the present study, we demonstrate that overexpression of IRF-3 significantly enhances virus-mediated transcription of the IFNA and IFNB genes in infected cells as well as IFN synthesis. IRF-3-mediated activation of IFN genes depends in part on carboxyl-terminal phosphorylation of a cluster of Ser/Thr residues, because a mutant with Ser/Thr to Ala substitutions activates the IFN promoter less efficiently. However, overexpression of IRF-3 in human 2FTGH cells alone results in the induction of an antiviral state, which depends on functional IFN signaling, because IRF-3 does not induce an antiviral state in mutant 2FTGH cells defective in either JAK-1 or p48 functions; also no antiviral effect of IRF-3 could be demonstrated in Vero cells that lack the IFNA and IFNB genes. This finding indicates that the observed antiviral activity of IRF-3 in 2FTGH cells results mainly from the induction of IFNs. Furthermore, E1A protein inhibited IRF-3-mediated stimulation of the IFNA4 promoter in transient expression assays; this inhibition could be reversed partially by overexpression of CBP/p300 and was not demonstrated with the mutant of E1A that does not bind p300. These results identify IRF-3 and CBP/p300 as integral components of the virus-induced complex that stimulates type 1 IFN gene transcription. The observation that adenovirus E1A antagonizes IRF-3 mediated activation suggests that E1A and IRF-3 may compete for binding to CBP/p300 and implicates a novel mechanism by which adenovirus may overcome the antiviral effects of the IFN pathway.
Figures

References
-
- Nguyen H, Hiscott J, Pitha P M. Cyt Growth Fact Rev. 1997;8:293–312. - PubMed
-
- Harada H, Fujita T, Miyamoto M, Kimura Y, Maruyama M, Furia A, Miyata T, Taniguchi T. Cell. 1989;58:729–739. - PubMed
-
- Vaughan P S, Aziz F, van Wijnen A J, Wu S, Harada H, Taniguchi T, Soprano K J, Stein J L, Stein G S. Nature (London) 1995;377:362–365. - PubMed
-
- Lin R, Mustafa A, Nguyen H, Hiscott J. J Biol Chem. 1994;269:17542–17549. - PubMed
-
- Weisz A, Marx P, Sharf R, Appella E, Driggers P H, Ozato K, Levi B-Z. J Biol Chem. 1992;267:25589–25596. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
