

The structural distribution of cooperative interactions in proteins: analysis of the native state ensemble
- PMID: 9707573
- PMCID: PMC21434
- DOI: 10.1073/pnas.95.17.9903
The structural distribution of cooperative interactions in proteins: analysis of the native state ensemble
Abstract
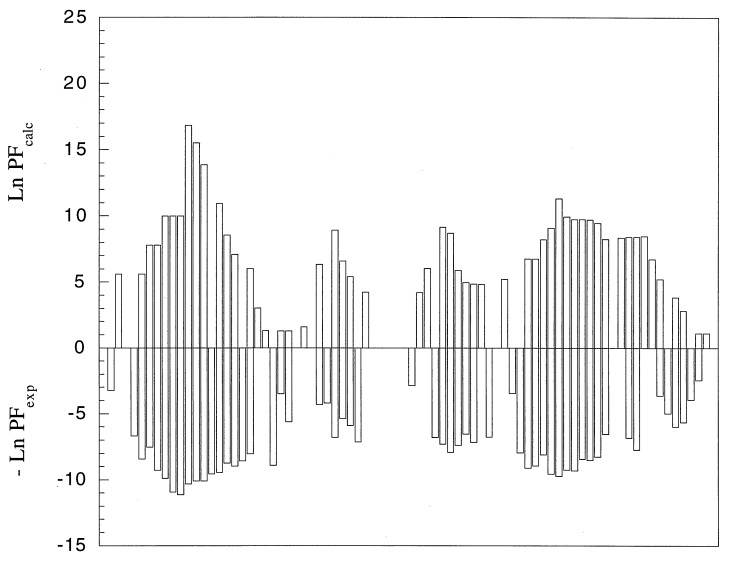
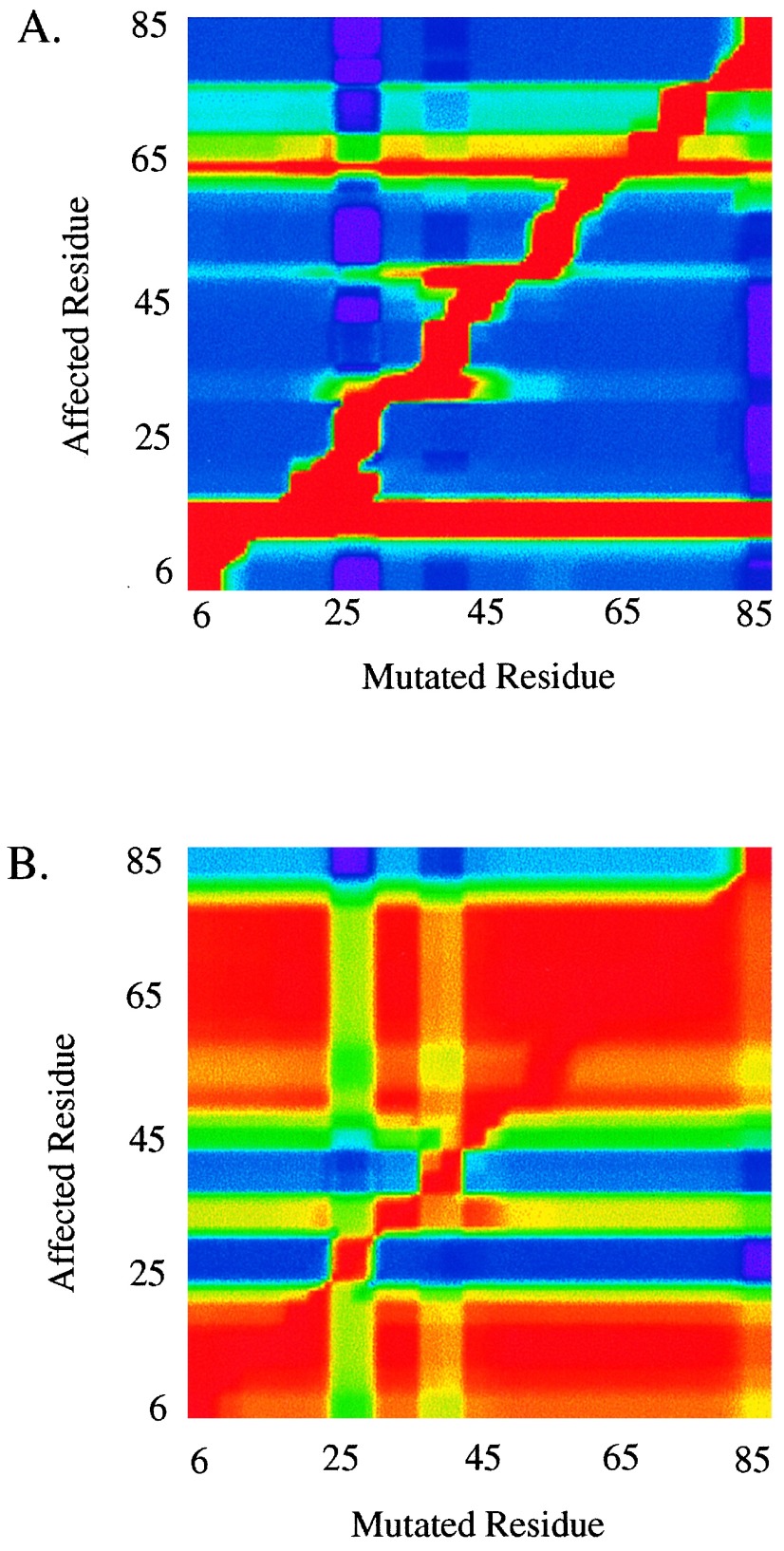
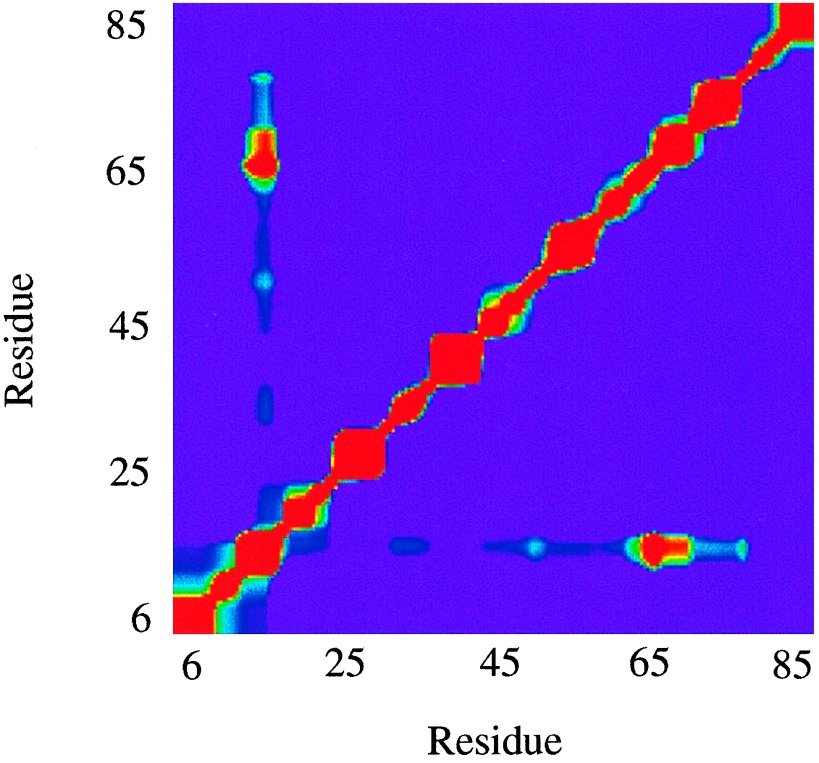
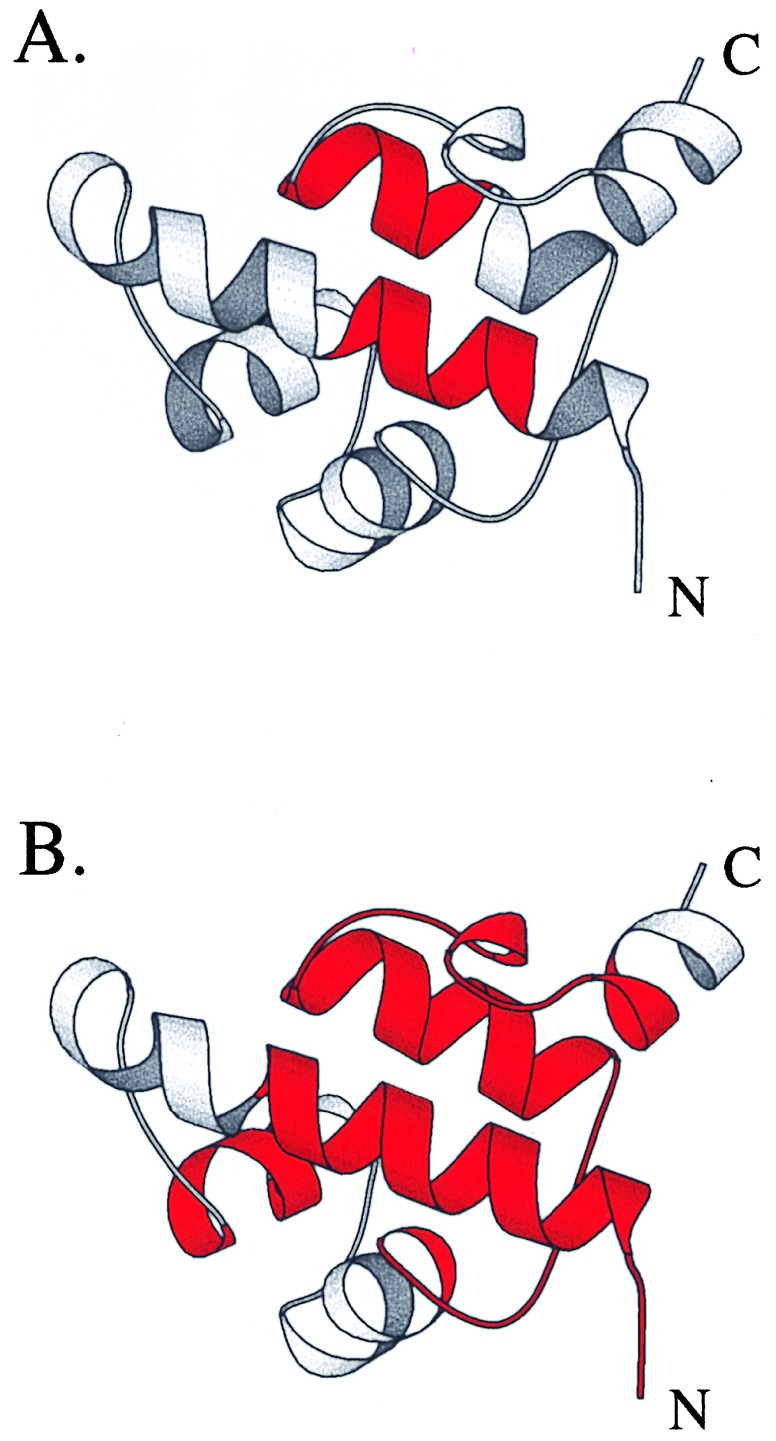
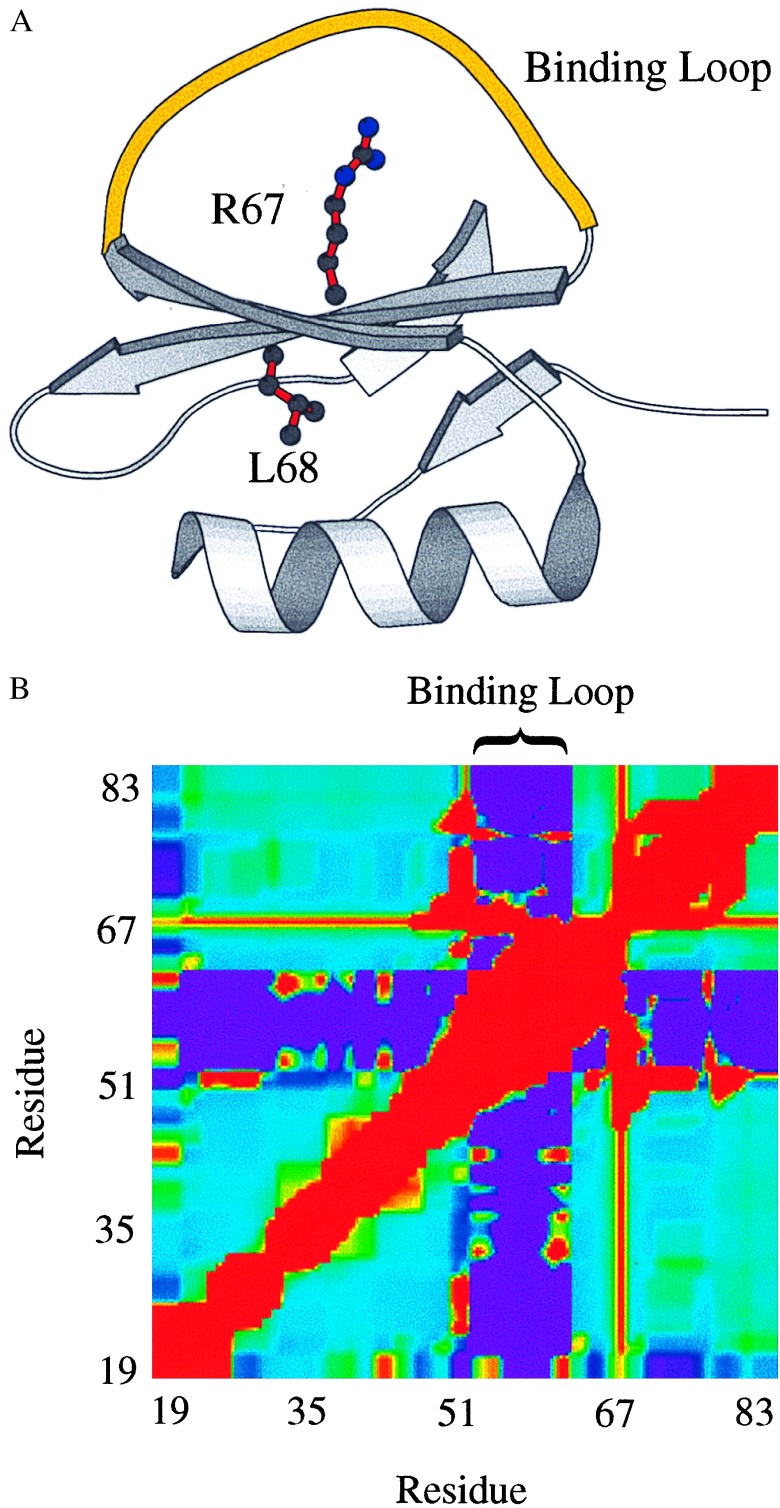
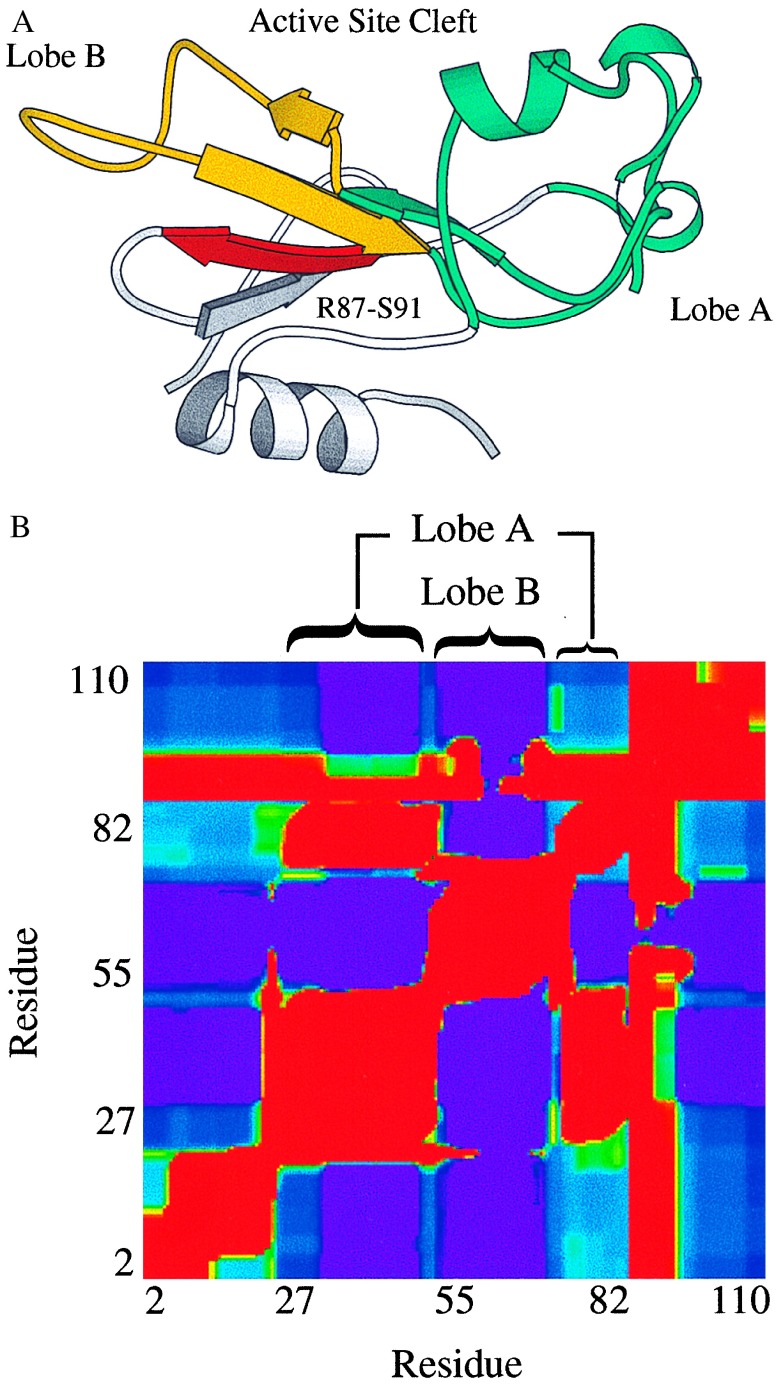
Cooperative interactions link the behavior of different amino acid residues within a protein molecule. As a result, the effects of chemical or physical perturbations to any given residue are propagated to other residues by an intricate network of interactions. Very often, amino acids "sense" the effects of perturbations occurring at very distant locations in the protein molecule. In these studies, we have investigated by computer simulation the structural distribution of those interactions. We show here that cooperative interactions are not intrinsically bi-directional and that different residues play different roles within the intricate network of interactions existing in a protein. The effect of a perturbation to residue j on residue k is not necessarily equal to the effect of the same perturbation to residue k on residue j. In this paper, we introduce a computer algorithm aimed at mapping the network of cooperative interactions within a protein. This algorithm exhaustively performs single site thermodynamic mutations to each residue in the protein and examines the effects of those mutations on the distribution of conformational states. The algorithm has been applied to three different proteins (lambda repressor fragment 6-85, chymotrypsin inhibitor 2, and barnase). This algorithm accounts well for the observed behavior of these proteins.
Figures
References
-
- Lumry R, Biltonen R, Brandts J F. Biopolymers. 1966;4:917–944. - PubMed
-
- Clarke J, Fersht A R. Fold Des. 1996;1:243–254. - PubMed
-
- Jeng M-F, Englander S W. J Mol Biol. 1991;221:1045–1061. - PubMed
-
- Radford S E, Buck M, Topping K D, Dobson C M, Evans P A. Proteins. 1992;14:237–248. - PubMed
-
- Loh S N, Prehoda K E, Wang J, Markley J L. Biochemistry. 1993;32:11022–11028. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
