

A single amino acid change in the acetylcholinesterase-like domain of thyroglobulin causes congenital goiter with hypothyroidism in the cog/cog mouse: a model of human endoplasmic reticulum storage diseases
- PMID: 9707574
- PMCID: PMC21435
- DOI: 10.1073/pnas.95.17.9909
A single amino acid change in the acetylcholinesterase-like domain of thyroglobulin causes congenital goiter with hypothyroidism in the cog/cog mouse: a model of human endoplasmic reticulum storage diseases
Abstract
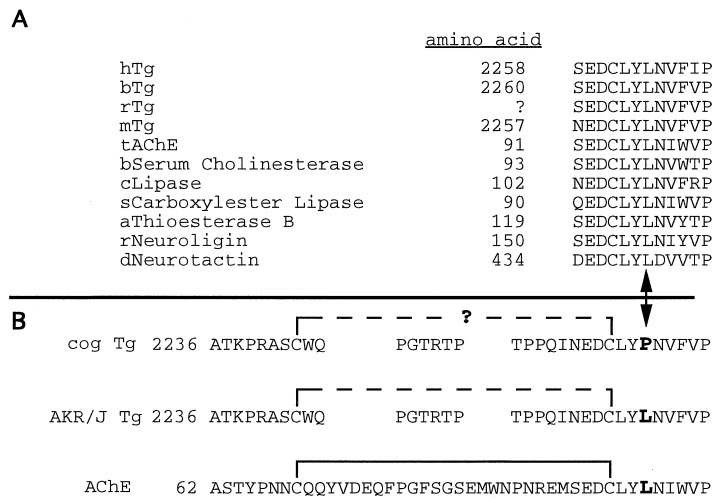
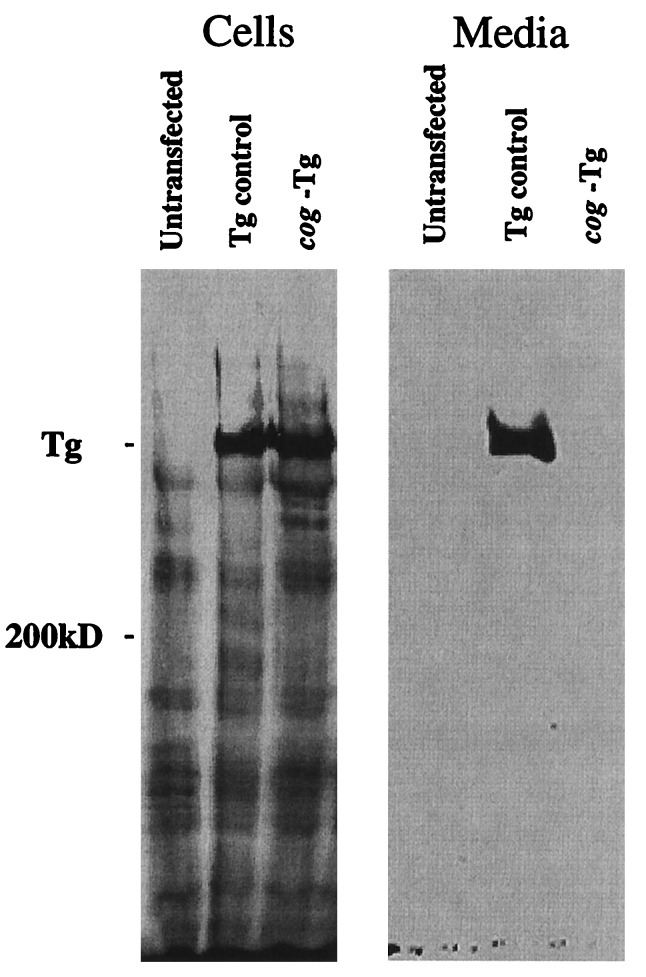
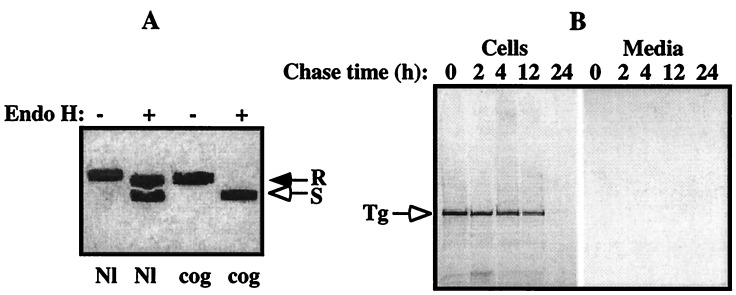
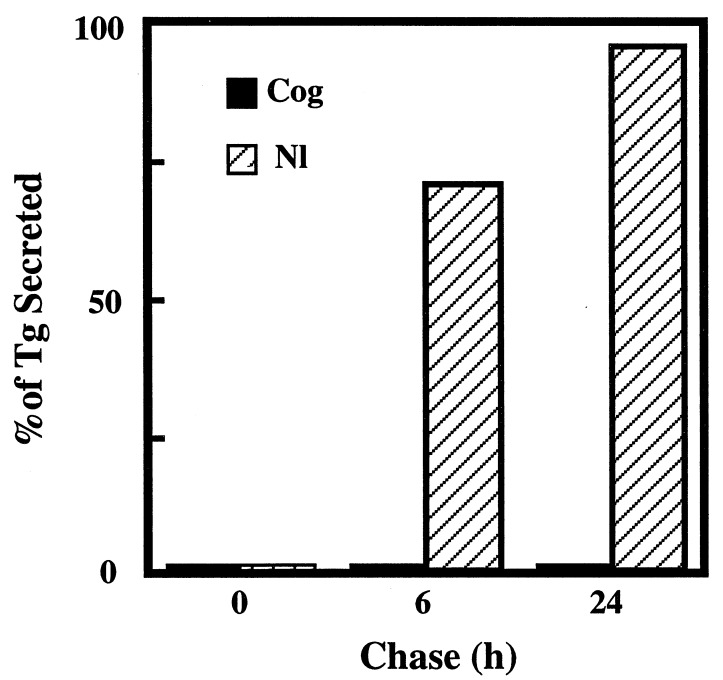
Newly synthesized thyroglobulin (Tg), the major secretory glycoprotein of the thyroid gland, folds and homodimerizes in the endoplasmic reticulum (ER) before its export to the site of iodination, where it serves as the precursor for thyroid hormone synthesis. In families with defective Tg export, affected individuals suffer from a thyroidal ER storage disease characterized by a distended thyrocyte ER containing misfolded Tg, along with induced ER molecular chaperones. Inherited as an autosomal recessive trait, deficient Tg causes congenital hypothyroidism in newborns that, if untreated, results in goiter along with serious cognitive and growth defects. Recently, a similar phenotype has been observed in inbred cog/cog mice, although the precise molecular defect has remained undefined. Here, we have isolated and cloned a full-length 8.5-kb Tg cDNA from cog/cog mice and unaffected isogenic AKR/J mice. Comparison of the complete sequences reveals that cog/cog mice express a Leu-2263 --> Pro missense mutation in the acetylcholinesterase-homology domain of Tg. Heterologous expression studies in COS cells indicate that cog Tg exhibits a severe defect in exit from the ER. Site-directed mutagenesis of cog Tg to convert the single amino acid back to Leu-2263 restores normal Tg secretion. We conclude that the cog mutation in Tg is responsible for this ER storage disease that causes thyroid dyshormonogenesis.
Figures

References
-
- Hammond C, Helenius A. Curr Opin Cell Biol. 1995;7:523–529. - PubMed
-
- Kim P S, Arvan P. Endocr Rev. 1998;19:173–202. - PubMed
-
- Ward C L, Omura S, Kopito R R. Cell. 1995;83:121–127. - PubMed
-
- Sifers R N, Finegold M J, Woo S L. Semin Liver Dis. 1992;12:301–310. - PubMed
-
- Prockop D J, Kivirikko K I. Annu Rev Biochem. 1995;64:403–434. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
